Zespół Noonana i podobne choroby. Zobacz, co oznacza „zespół Noonana” w innych słownikach.
E.V. Tozliyan, endokrynolog dziecięcy, genetyk, dr, Separate podział strukturalny„Badawczy Kliniczny Instytut Pediatrii” Państwowa Budżetowa Instytucja Edukacyjna Wyższego Kształcenia Zawodowego Rosyjski Narodowy Uniwersytet Medyczny im. NI Ministerstwo Zdrowia Federacji Rosyjskiej Pirogowa, Moskwa Słowa kluczowe: dzieci, zespół Noonana, diagnostyka.
Słowa kluczowe: dzieci, zespół Noonana, diagnostyka.
W artykule opisano zespół Noonana (zespół Ullricha-Noonana, zespół turneroidowy z prawidłowym kariotypem) – rzadką wrodzona patologia, dziedziczony w sposób autosomalny dominujący, niesie charakter rodzinny Zdarzają się jednak także sporadyczne przypadki. Zespół zakłada obecność fenotypu charakterystycznego dla zespołu Shereshevsky'ego-Turnera u kobiet i mężczyzn o prawidłowym kariotypie. Przedstawione przez obserwacja kliniczna. Trudności w poszukiwaniach diagnostyki różnicowej i brak świadomości lekarzy nt ten syndrom oraz znaczenie podejścia interdyscyplinarnego.
Fakt historyczny
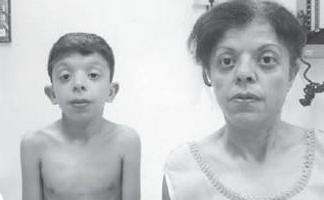
Po raz pierwszy o niezwykły syndrom wzmiankowany przez O. Kobylińskiego w 1883 r. (fot. 1).
Najstarszy znany przypadek kliniczny Zespół Noonana, opisany w 1883 r. przez O. Kobylińskiego
Choroba została opisana w 1963 roku przez amerykańską kardiolog Jacqueline Noonan, która opisała dziewięciu pacjentów ze zwężeniem zastawki tętnica płucna, niski wzrost, hiperteloryzm, umiarkowany spadek inteligencji, opadanie powiek, wnętrostwo i nieprawidłowości szkieletowe. Doktor Noonan, który praktykował jako kardiolog dziecięcy na Uniwersytecie Iowa zauważył, że dzieci z rzadki typ wada serca - zwężenie zastawki płucnej - często obserwowano typowe nieprawidłowości fizyczne, takie jak niski wzrost, skrzydlata szyja, szeroko osadzone oczy i nisko osadzone uszy. W równym stopniu dotknięci byli chłopcy i dziewczęta. Doktor John Opitz, były student Noonan jako pierwszy ukuł termin „zespół Noonana” na określenie stanu dzieci wykazujących objawy podobne do tych opisanych przez Noonana. Noonan napisał później artykuł „Hiperteloryzm z fenotypem Turnera”, a nazwa „zespół Noonana” została oficjalnie uznana w 1971 r. na sympozjum poświęconym chorobom sercowo-naczyniowym.
Etiologia i patogeneza
Zespół Noonana jest chorobą autosomalną dominującą o zmiennej ekspresji (ryc. 1). Gen zespołu Noonana jest zlokalizowany w długie ramię chromosom 12. Nie można wykluczyć heterogeniczności genetycznej zespołu. Opisano sporadyczne i rodzinne postaci zespołu z autosomalną dominującą formą dziedziczenia. W sprawach rodzinnych zmutowany gen jest dziedziczony z reguły po matce, ponieważ z powodu poważnych wad rozwojowych układ moczowo-płciowy mężczyźni cierpiący na tę chorobę są często bezpłodni. Większość zgłaszanych przypadków ma charakter sporadyczny i jest spowodowana mutacjami de novo.
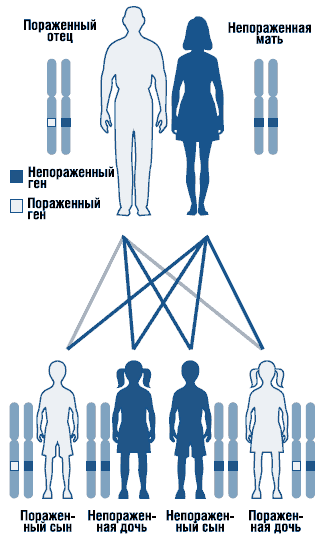
. Autosomalny dominujący typ dziedziczenia
Opisane kombinacje zespołu Noonana z nerwiakowłókniakowatością typu I w kilku rodzinach zasugerowały możliwy związek pomiędzy dwoma niezależnymi loci 17q11.2 chromosomu 17. U niektórych pacjentów mikrodelecje wykrywane są w locus 22q11 chromosomu 22; w tych przypadkach objawy kliniczne Zespół Noonana łączy się z niedoczynnością grasicy i zespołem DiGeorge'a. Wielu autorów omawia udział domniemanych genów limfogenezy w patogenezie zespołu ze względu na obecność anomalii twarzowych i somatycznych podobnych do zespołu Turnera oraz wysoką częstość patologii system limfatyczny.
Bardzo powszechny powód Zespół Noonana to mutacja genu PTPN11, która występuje u około 50% pacjentów. Białko kodowane przez gen PTPN11 należy do rodziny cząsteczek regulujących odpowiedź komórek eukariotycznych na sygnały zewnętrzne. Największa liczba mutacje w zespole Noonan zlokalizowane są w eksonach 3, 7 i 13 genu PTPN11, kodującego domeny białkowe odpowiedzialne za przejście białka do stanu aktywnego.
Możliwe pomysły na patogenezę są reprezentowane przez następujące mechanizmy:
Szlak RAS-MAPK jest bardzo ważnym szlakiem przekazywania sygnału, poprzez który ligandy zewnątrzkomórkowe - pewne czynniki wzrost, cytokiny i hormony - stymulują proliferację, różnicowanie, przeżycie i metabolizm komórek (ryc. 2). Po związaniu liganda receptory na powierzchni komórki ulegają fosforylacji w miejscach w ich regionie endoplazmatycznym. Wiązanie to obejmuje białka adaptorowe (np. GRB2), które tworzą konstytutywny kompleks z czynnikami wymiany nukleotydów guaninowych (np. SOS), które przekształcają nieaktywny RAS związany z PKB w jego aktywną postać związaną z GTP. Aktywowane białka RAS następnie aktywują kaskadę RAF-MEKERK poprzez serię reakcji fosforylacji. W rezultacie aktywowana ERK wchodzi do jądra, aby zmienić transkrypcję docelowych genów i dostosowuje aktywność celów endoplazmatycznych, aby wywołać odpowiednią krótko- i długoterminową odpowiedź komórkową na bodziec. Wszystkie geny zaangażowane w zespół Noonana kodują białka integralne z tym szlakiem oraz mutacje powodujące chorobę, zwykle wzmacniają sygnał przechodzący tą ścieżką.
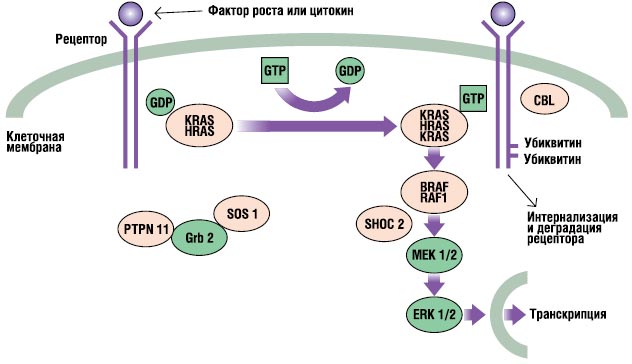
. Szlak sygnalizacyjny RAS-MAPK. Sygnały wzrostu przekazywane są z receptorów aktywowanych czynnikiem wzrostu do jądra. Mutacje w PTPN11, KRAS, SOS1, NRAS i RAF1 są powiązane z zespołem Noonan, a mutacje w SHOC2 i CBL są powiązane z fenotypem podobnym do zespołu Noonan
Charakterystyka kliniczna zespołu Noonana
Fenotyp pacjentów z zespołem Noonana przypomina zespół Turnera: krótka szyja z fałdem skrzydłowym lub niskim wzrostem włosów, niskim wzrostem, hiperteloryzmem szpar powiekowych (zdjęcie 2). Mikroanomalie twarzy obejmują antymongoloidalne szpary powiekowe, opadające kąciki zewnętrzne, opadanie powiek, nakąt, nisko osadzone przedsionki, zwiniętą helisę uszy, wada zgryzu, rozszczep języczka podniebienie miękkie, Podniebienie gotyckie, mikrognacja i mikrogenia. Klatka piersiowa ma kształt tarczycy z hipoplastycznymi, szeroko rozstawionymi sutkami, mostek wystaje w górnej części i opada w dolnej. Około 20% pacjentów ma umiarkowaną patologię szkieletu. Najczęstszym rodzajem deformacji jest deformacja w kształcie lejka. klatka piersiowa, kifoza, skolioza; rzadziej - zmniejszenie liczby kręgów szyjnych i ich zespolenie, przypominające anomalie w zespole Klippla-Feila.
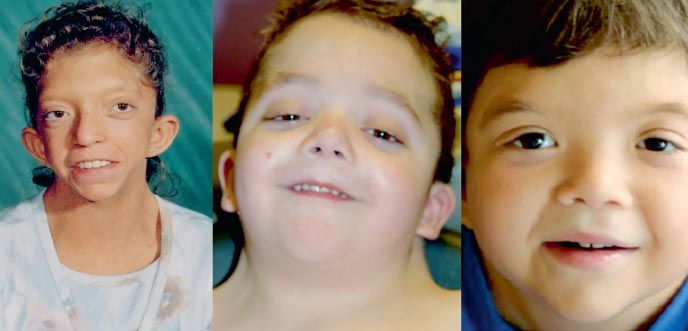
. Fenotypy zespołu Noonana
Pacjenci z zespołem Noonana mają zwykle blond, gęste, kręcone włosy z nietypowym wzrostem na czubku głowy; ciemne miejsca na skórze, nadmierne owłosienie, dystrofia płytek paznokciowych, nieprawidłowości w wyrzynaniu i położeniu zębów, skłonność do powstawania blizn keloidowych, zwiększona rozciągliwość skóry. U jednej trzeciej pacjentów występuje obrzęk limfatyczny obwodowy, częściej u dzieci występuje obrzęk limfatyczny dłoni i stóp młodym wieku. Częstym objawem jest patologia wzroku (krótkowzroczność, zez, umiarkowany wytrzeszcz itp.). Opóźnienie wzrostu występuje u około 75% pacjentów, jest bardziej widoczne u chłopców i zwykle jest nieistotne. Opóźnienie wzrostu objawia się w pierwszych latach życia, rzadziej odnotowuje się niewielkie deficyty wzrostu i masy ciała przy urodzeniu. Od pierwszych miesięcy życia następuje spadek apetytu. Wiek kostny zwykle pozostaje w tyle za wiekiem paszportowym.
Cechą charakterystyczną tego zespołu jest jednostronne lub obustronne wnętrostwo, które występuje u 70–75% mężczyzn, u dorosłych azoospermia, oligospermia, zmiany zwyrodnieniowe jądra. Niemniej jednak dojrzewanie następuje samoistnie, czasami z pewnym opóźnieniem. Dziewczęta często doświadczają opóźnienia w powstawaniu miesiączki, a czasami występują nieprawidłowości cykl miesiączkowy. Płodność może być prawidłowa u pacjentów obu płci.
U ponad połowy pacjentów stwierdza się upośledzenie umysłowe, zwykle niewielkie. Często obserwuje się osobliwości behawioralne, rozhamowanie i zaburzenia deficytu uwagi. Mowa jest zwykle lepiej rozwinięta niż inne sfery intelektualne. Stopień spadku inteligencji nie koreluje z nasileniem zaburzeń somatycznych [Marincheva G.S., 1988]. W pojedynczych przypadkach wady rozwojowe ośrodkowego system nerwowy(wodogłowie, rozszczep kręgosłupa), zakrzepowo-zatorowy zawał mózgu, prawdopodobnie związany z hipoplazją naczyń.
Wady narządy wewnętrzne z zespołem Noonana są dość charakterystyczne. Najbardziej typowe są anomalie sercowo-naczyniowe: zwężenie zastawki tętnicy płucnej (około 60% chorych), Kardiomiopatia przerostowa(20–30%), nieprawidłowości strukturalne zastawka mitralna, ubytki przegrody międzyprzedsionkowej, tetralogia Fallota; Koarktację aorty opisano wyłącznie u mężczyzn.
U jednej trzeciej pacjentów rejestruje się wady układu moczowego (niedorozwój nerek, zduplikowanie miednicy, wodonercze, moczowód olbrzymi itp.).
Dość często w przypadku zespołu Noonana występuje zwiększone krwawienie, szczególnie gdy interwencje chirurgiczne V Jama ustna i nosogardzieli. Wykrywane są różne zaburzenia krzepnięcia: niewydolność układu płytek krwi, obniżony poziom czynników krzepnięcia, zwłaszcza XI i XII, wydłużony czas tromboplastyny. Istnieją doniesienia o połączeniu zespołu Noonana z białaczką i mięśniakomięsakiem prążkowanokomórkowym, co może wskazywać na niewielki wzrost ryzyka nowotworu złośliwego u tych pacjentów.
W tabeli 1 przedstawiono cechy fenotypu zespołu Noonana, które zmieniają się wraz z wiekiem pacjenta. Tabela 2 przedstawia korelację między fenotypem a genotypem w zespole Noonana.
Tabela 1. Typowe rysy twarzy pacjentów z zespołem Noonana według wieku
| Czoło, twarz, włosy | Oczy | Uszy | Nos | Usta | Szyja | |
| Nowo narodzony* | Wysokie czoło, niska linia włosów z tyłu głowy | Hiperteloryzm, opadający szpary powiekowe, fałd epikantu | – | Korzeń krótki i szeroki, zagłębiony, wierzchołek zadarty | Głęboko wpuszczona rynienka podnosowa, wysokie szerokie szczyty czerwonej granicy warg, mikrognacja | Nadmiar skóry z tyłu głowy |
| Niemowlę (2–12 miesięcy) | Duża głowa, wysokie i wystające czoło | Hiperteloryzm, opadanie powiek lub grube opadające powieki | – | Korzeń wklęsły krótki i szeroki | – | – |
| Dziecko (1–12 lat) | Szorstkie rysy, pociągła twarz | – | – | – | – | – |
| Nastolatki (12–18 lat) | Miopatyczna twarz | – | – | Most jest wysoki i cienki | – | Widoczne powstawanie fałdów szyjnych |
| Dorosły (> 18 lat) | Charakterystyczne rysy twarzy są wyrafinowane, skóra wydaje się cienka i przezroczysta | – | – | Wystający fałd nosowo-wargowy | – | – |
| Wszystkich grup wiekowych | – | Niebieskie i zielone tęczówki, brwi w kształcie rombu | Uszy niskie, odwrócone do tyłu, z grubymi fałdami | – | – | – |
Tabela 2. Korelacje między genotypem a fenotypem w zespole Noonana*
| Układ sercowo-naczyniowy | Wysokość | Rozwój | Skóra i włosy | Inny | |
| PTPN11 (około 50%) | Zwężenie pnia płucnego jest bardziej wyraźne; mniej – kardiomiopatia przerostowa i ubytek przegrody międzyprzedsionkowej | Mniejsza wysokość; niższe stężenie IGF1 | Pacjenci z N308D i N308S mają lekko obniżoną lub normalną inteligencję | – | Bardziej nasilona jest skaza krwotoczna i młodzieńcza białaczka mielomonocytowa |
| SOS1 (około 10%) | Mniejszy ubytek przegrody międzyprzedsionkowej | Wyższy wzrost | Mniejszy spadek inteligencji, opóźniony rozwój mowy | Podobny do zespołu sercowo-skórno-twarzowego | – |
| RAF1 (około 10%) | Cięższa kardiomiopatia przerostowa | – | – | Więcej znamiona, soczewica, kawiarnia z mlekiem | – |
| KRAS (<2%) | – | – | Poważniejsze opóźnienie poznawcze | Podobny do zespołu sercowo-skórno-twarzowego | – |
| NRAS (<1%) | – | – | – | – | – |
Dane z badań laboratoryjnych i funkcjonalnych
Nie ma specyficznych markerów biochemicznych do diagnozowania zespołu Noonana. U niektórych pacjentów stwierdza się zmniejszenie spontanicznego nocnego wydzielania hormonu wzrostu przy prawidłowej odpowiedzi na farmakologiczne testy stymulujące (klonidynę i argininę), zmniejszenie poziomu somatomedyny-C i zmniejszenie odpowiedzi somatomedyn na podanie hormon wzrostu.
Kryteria diagnozy
Rozpoznanie zespołu Noonana stawia się na podstawie objawów klinicznych, w niektórych przypadkach rozpoznanie potwierdzają wyniki molekularnego badania genetycznego. Kryteriami rozpoznania zespołu są obecność charakterystycznej twarzy (z prawidłowym kariotypem) w połączeniu z jednym z następujących objawów: patologia serca, niski wzrost lub wnętrostwo (u chłopców), opóźnione dojrzewanie (u dziewcząt). Aby zidentyfikować patologię sercowo-naczyniową, konieczne jest przeprowadzenie badania ultrasonograficznego serca z dynamicznym określeniem wielkości jam i ściany komór. Diagnostyka prenatalna choroby jest możliwa dzięki monitorowaniu ultrasonograficznemu, które pozwala wykryć wady serca i nieprawidłowości w budowie szyi.
Diagnostyka różnicowa
U dziewcząt w diagnostyce różnicowej rozpoznaje się przede wszystkim zespół Turnera; Badanie cytogenetyczne może wyjaśnić diagnozę. Objawy fenotypowe zespołu Noonana występują w wielu innych chorobach: zespole Williamsa, zespole LEOPARDA, zespole Dubowitza, zespole sercowo-twarzowo-skórnym, Cornelii de Lange, Cohena, Rubinsteina-Taybiego itp. Dokładna identyfikacja tych chorób będzie możliwa jedynie poprzez przeprowadzenie badań molekularnych badania genetyczne każdego zespołu z istotnym materiałem klinicznym, który jest obecnie aktywnie rozwijany.
Leczenie
Leczenie pacjentów z zespołem Noonana ma na celu wyeliminowanie wad układu sercowo-naczyniowego, normalizację funkcji psychicznych, stymulację wzrostu i rozwoju seksualnego. W leczeniu pacjentów z dysplazją zastawek płucnych z powodzeniem stosuje się między innymi walwuloplastykę balonową. Aby pobudzić rozwój umysłowy, stosuje się leki nootropowe i naczyniowe. Leki stymulujące rozwój seksualny wskazane są głównie u pacjentów z wnętrostwem. Preparaty ludzkiej gonadotropiny kosmówkowej stosuje się w dawkach dostosowanych do wieku. W starszym wieku – przy hipogonadyzmie – preparaty testosteronu. W ostatnich latach w leczeniu pacjentów z zespołem Noonana zaczęto stosować rekombinowane formy ludzkiego hormonu wzrostu. Dane kliniczne potwierdza wzrost poziomu somatomedyny-C i specyficznie wiążącego białka w trakcie terapii. Ostateczny wzrost pacjentów długotrwale leczonych hormonem wzrostu przekracza w niektórych przypadkach średni wzrost członków rodziny.
Prognoza na całe życie zależy od ciężkości patologii sercowo-naczyniowej.
Zapobieganie choroby opiera się na danych uzyskanych z medycznego poradnictwa genetycznego.
Medyczne poradnictwo genetyczne
Prowadząc medyczną poradnię genetyczną, należy kierować się autosomalnym dominującym typem dziedziczenia i wysokim (50%) ryzykiem nawrotu choroby w rodzinie z postaciami dziedzicznymi. Aby określić charakter rodzaju dziedziczenia, konieczne jest dokładne badanie rodziców, ponieważ zespół może objawiać się minimalnymi objawami klinicznymi. Obecnie rozwija się i udoskonala molekularną diagnostykę genetyczną choroby poprzez typowanie mutacji w genach: PTPN11, SOS1, RAF1, KRAS, NRAS itp. Trwają prace nad metodami diagnostyki prenatalnej choroby.
Obserwacja kliniczna
Chłopiec G., lat 9 (zdjęcie 3), był obserwowany w miejscu zamieszkania przez genetyka z rozpoznaniem „patologii chromosomalnej?, zespołu Williamsa (specyficzny fenotyp, pogrubienie płatków zastawki mitralnej, hiperkalcemia raz na 3 lata) ?.
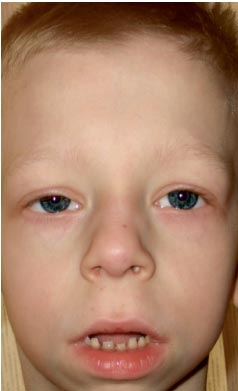
. Cechy fenotypu dziecka z zespołem Noonana (wydłużony szkielet twarzy z „pulchnymi policzkami”, krótka szyja, fałdy skrzydłowe na szyi, skrócony nos z nozdrzami otwartymi do przodu, pulchne wargi, opadający podbródek, antymongoloidalne nacięcie szpary powiekowej , wady zgryzu, makrostomia)
Uskarżanie się na zmniejszoną pamięć, zmęczenie, zmniejszone tempo wzrostu.
Historia rodzinna : rodzice są Rosjanami ze względu na narodowość, niespokrewnieni i nie mają ryzyka zawodowego, zdrowi. Wzrost ojca 192 cm, wzrost matki 172 cm W rodowodzie nie było przypadków chorób psychicznych, epilepsji ani opóźnień rozwojowych.
Historia życia i choroby : chłopiec z drugiej ciąży (pierwsza ciąża - m/a), która przez cały czas przebiegała z zagrożeniem poronieniem, któremu towarzyszyło wielowodzie. Pierwszy poród, terminowy, szybki, masa urodzeniowa – 3400 g, długość – 50 cm, od razu krzyknął, Apgar – 7/9 pkt. Przy porodzie neonatolog zwrócił uwagę na nietypowy fenotyp dziecka i zalecił badanie kariotypu, wynik wyniósł 46,XY (prawidłowy kariotyp męski). Podejrzewano wrodzoną niedoczynność tarczycy, wykonano badanie profilu tarczycy, w wyniku którego stwierdzono prawidłowy stan tarczycy. Następnie dziecko było obserwowane przez genetyka z przypuszczalną diagnozą zespołu Williamsa. Wczesny okres poporodowy jest pozbawiony cech charakterystycznych. Rozwój motoryczny według wieku, pierwsze słowa – o rok, mowa frazowa – w wieku 2 lat i 3 miesięcy.
W wieku 8 lat został skonsultowany przez endokrynologa w związku ze zmniejszonym tempem wzrostu, zmęczeniem i zmniejszoną pamięcią. Badanie rentgenowskie dłoni wykazało umiarkowane opóźnienie wieku kostnego (BA) w stosunku do wieku paszportowego (BA odpowiadało 6 latom). Badanie profilu tarczycy wykazało umiarkowany wzrost hormonu tyreotropowego przy prawidłowym poziomie wolnego T4 i innych wskaźników; USG tarczycy - bez patologii. Zalecono terapię hormonalną i późniejszą obserwację dynamiczną.
Biorąc pod uwagę niepewność diagnozy w miejscu zamieszkania, genetyk wysłał dziecko do Moskiewskiego Regionalnego Centrum Konsultacyjno-Diagnostycznego dla Dzieci w celu wyjaśnienia diagnozy.
Obiektywne dane badawcze:
Wzrost – 126 cm, waga – 21 kg.
Rozwój fizyczny jest poniżej średniej, harmonijny. Wzrost Sds odpowiada –1 (norma – –2+2). Cechy fenotypowe (zdjęcie 3): wydłużony szkielet twarzy z „pulchnymi policzkami”, krótka szyja, fałdy skrzydłowe na szyi, niski porost włosów na szyi, skrócony nos z nozdrzami otwartymi do przodu, pulchne usta, opadający podbródek, antymongoloidalne nacięcie szpary powiekowe, wady zgryzu, makrostomia, hiperteloryzm sutków, asymetria klatki piersiowej, niepełny syndyktylia skórna II–III palca stopy, silna nadmierna ruchliwość stawów międzypaliczkowych, łamliwe, suche paznokcie. Narządy wewnętrzne – bez żadnych osobliwości. Rozwój płciowy – Tanner I (co odpowiada okresowi przedpokwitaniowemu).
Dane z badań laboratoryjnych i funkcjonalnych:
Analiza kliniczna krwi i moczu jest prawidłowa.
Biochemiczne badanie krwi - wskaźniki mieszczą się w normalnych granicach.
Profil tarczycy (TSH) – 7,5 µIU/ml (w normie – 0,4–4,0), pozostałe wskaźniki w normie.
Hormon somatotropowy (GH) – 7 ng/ml (w normie – 7–10), somatomedyna-C – 250 ng/ml (w normie – 88–360).
USG tarczycy - bez patologii.
USG narządów wewnętrznych - bez żadnych cech.
EKG – częstoskurcz zatokowy, prawidłowe położenie osi elektrycznej serca.
EchoCG – MVP I stopnia z minimalną niedomykalnością, śluzakowym pogrubieniem płatków zastawki mitralnej, dodatkowym cięciwą w jamie lewej komory.
R-grafia kręgosłupa – skolioza prawostronna odcinka piersiowego kręgosłupa I stopień.
R-grafia dłoni z chwytem przedramion – wiek kostny 7–8 lat.
Nie zarejestrowano żadnych wzorców aktywności padaczkowej w EEG.
MRI mózgu – brak zmian patologicznych.
Audiogram – bez patologii.
Diagnostyka DNA: badanie genetyki molekularnej - nie wykryto delecji w badanych loci regionu krytycznego 7 chromosomu; W 11. eksonie genu SOS1 wykryto mutację Gly434Ary (1230G>A) (analiza genu PTPN11 – nie stwierdzono mutacji), co jest charakterystyczne dla zespołu Noonan.
Konsultacje specjalistyczne:
Endokrynolog– subkliniczna niedoczynność tarczycy, niepełna kompensacja leku.
Okulista– astygmatyzm.
Neurolog– dystonia wegetatywno-naczyniowa. Reakcje neurotyczne.
Kardiolog– kardiopatia czynnościowa.
Chirurg ortopeda- zła postawa. Deformacja klatki piersiowej.
Genetyk– Zespół Noonana.
Biorąc pod uwagę fenotyp dziecka, historię choroby oraz wyniki badań dodatkowych, postawiono rozpoznanie zespołu Noonana, co potwierdzono wynikiem badania genetyki molekularnej.
Zatem przedstawiona obserwacja kliniczna pokazuje trudności w diagnostyce różnicowej, potrzebę włączenia poszczególnych objawów w ogólny fenotyp konkretnego stanu patologicznego w celu ukierunkowanej, terminowej diagnozy poszczególnych postaci chorób dziedzicznych oraz znaczenie molekularnych metod genetycznych dla wyjaśnienia diagnoza. Wczesne rozpoznanie i wyjaśnienie genezy każdego zespołu jest szczególnie ważne, ponieważ pozwala znaleźć optymalne podejście do leczenia tych schorzeń i zapobiegania możliwym powikłaniom (aż do niepełnosprawności dziecka włącznie); zapobieganie nawrotom chorób dziedzicznych w dotkniętych rodzinach (poradnictwo lekarskie i genetyczne). Narzuca to potrzebę jasnego poruszania się przez lekarzy różnych specjalności w przepływie dziedzicznie zdeterminowanej patologii.
Bibliografia:
- Baird P., Zespół De Jonga B. Noonana (fenotyp XX i XY Turnera) w trzech pokoleniach rodziny // J. Pediatr., 1972, tom. 80, s. 110–114.
- Hasegawa T., Ogata T. i in. Koarktacja aorty i hupoplazja nerek u chłopca z anomaliami powierzchni Turnera/Noonana i kariotypem 46, XY: model kliniczny możliwego upośledzenia przypuszczalnego genu(ów) limfogennego(-ych) dla stygmatów somatycznych Turnera // Hum. Genet., 1996, tom. 97, r. 564–567.
- Fedotova T.V., Kadnikova V.A. i in. Kliniczna i molekularna analiza genetyczna zespołu Noonana. Materiały VI Kongresu Rosyjskiego Towarzystwa Genetyki Medycznej. Genetyka medyczna, suplement nr 5, 2010, s. 184.
- Ward K.A., Moss C., McKeown C. Zespół sercowo-twarzowo-skórny: przejaw zespołu Noonana? // br. J. Dermatol., 1994, tom. 131, s. 270–274.
- Municchi G., Pasquino A.M. i in. Leczenie hormonem wzrostu w zespole Noonana: opis czterech przypadków, które osiągnęły ostateczny wzrost // Horm. Res., 1995, tom. 44, r. 164–167.
U dzieci występują różne wrodzone patologie. Niektóre z nich są uważane za dość powszechne. Ale są też choroby rzadkie. Ich rozpowszechnienie jest niskie. Jednak wiele z nich ma dość poważne konsekwencje. Jedną z tych patologii jest zespół Noonana. Przyjrzyjmy się temu bardziej szczegółowo w dalszej części. W artykule opisano, jak objawia się zespół Noonana i dlaczego się pojawia. W tekście zaprezentowane zostanie także zdjęcie choroby.
Informacje ogólne
Zespół Noonana jest patologią dziedziczną. Zmutowany gen PTPN11 jest przekazywany potomstwu od rodziców, którzy są jego nosicielami. Z reguły mężczyźni są bezpłodni. Dlatego gen jest przekazywany przez linię matczyną. Zwykle zwraca się uwagę na rodzinny charakter tej choroby. Rzadkie przypadki niezwiązane z dziedzicznością, ale są również rejestrowane w praktyce.
Odniesienie historyczne
Pewnego razu Jacqueline Noonan, która prowadziła praktykę kardiologa dziecięcego, pracując w klinice Uniwersytetu Iowa, zauważyła, że pewna grupa dzieci, do której zaliczali się zarówno chłopcy, jak i dziewczęta cierpiące na zwężenie tętnicy płucnej, często wyróżniała się niskim wzrostem, szeroko rozstawione oczy i błoniaste szyje, nisko położone uszy i opadanie powiek. Po zbadaniu połączenia objawów chorób serca z innymi anomaliami rozwojowymi u 833 pacjentów lekarz napisał artykuł w 1962 roku. W swojej pracy opisała dziewięć przypadków, w których na tle typu wrodzonego odnotowano charakterystyczne rysy twarzy i niski wzrost. Chorobę stwierdzono zarówno u mężczyzn, jak iu kobiet. 
Zespół Noonana: objawy
Istnieje wiele charakterystycznych cech, które odróżniają patologię od innych. Należą do nich w szczególności:
- Niski wzrost. Dla kobiet - 1,53, dla mężczyzn - 1,63 m. W chwili urodzenia zarówno długość ciała, jak i masa dziecka mieszczą się w normalnych granicach. Karłowatość rozpoczyna się w wieku 2-3 lat.
- Zmiany twarzy. Pacjenci, u których zdiagnozowano zespół Noonana, mają szeroko rozstawione formy. W wewnętrznym kąciku znajduje się fałd skórny. Opadanie powiek występuje również na skutek osłabienia pracy mięśni lub małych oczodołów. Około dwie trzecie wszystkich dzieci z patologią ma opadanie powiek i rozwija się zez.
- Zaburzenia w budowie szczęk. Górna jest słabo rozwinięta, obserwuje się łukowate podniebienie wysokie. Na obu szczękach względne położenie zębów jest nieprawidłowe.
- Niskie dopasowanie lub deformacja uszu. W wyniku tej anomalii słuch jest upośledzony.
- Krótka i szeroka szyja.
- Klatka piersiowa w kształcie tarczy z szeroko rozstawionymi sutkami.
- Deformacja stawu łokciowego (wrodzona).
- Płaskostopie.
- Krótkie palce.
Wady rozwojowe narządów wewnętrznych
Zespołowi Noonana najczęściej towarzyszą zaburzenia w funkcjonowaniu układu sercowo-naczyniowego. Na tle patologii ujawnia się zwężenie (zwężenie) pnia płucnego, a także ubytek przegrody międzykomorowej. Drugie miejsce zajmują anomalie układu moczowo-płciowego. W ten sposób wykrywane są wady nerek, takie jak hipoplazja (niepowodzenie rozwoju tkanki) lub brak jednej nerki. Jeśli chodzi o dojrzewanie, waha się ono od normalnego do całkowicie wadliwego. U dziewcząt często miesiączka pojawia się późno, u chłopców występuje wnętrostwo lub całkowity brak jąder. Wykrywane są także zaburzenia spermatogenezy. W niektórych przypadkach plemniki są całkowicie nieobecne. Podczas operacji zespołu Noonana obserwuje się zwiększone krwawienie. U niektórych pacjentów mogą również występować łagodne nieprawidłowości. 
Formularze
Istnieją dwa rodzaje patologii:
- Formularz rodzinny. Wyróżnia się dziedziczną naturą typu autosomalnego dominującego. Nosiciele zmutowanego genu mają potomstwo z patologią.
- Sporadyczna forma. W tym przypadku mutacja pojawia się od przypadku do przypadku. Nie zidentyfikowano jednak żadnego czynnika dziedzicznego.
Powoduje
Najczęstszym czynnikiem powodującym patologię jest mutacja w genie PTPN11. Przyczynę tę stwierdza się u 50% pacjentów. Jednak u pewnego odsetka osób z tym zespołem czynnik genetyczny jest niejasny. Dziedziczenie patologii występuje w formie autosomalnej dominującej. Zespół może być wywołany nową mutacją. Wynika z tego, że rodzice posiadający ten gen nie mają dużych szans na posiadanie kolejnego dziecka.
Diagnoza
Montuje się go zgodnie z charakterystycznymi znakami zewnętrznymi (opisanymi powyżej). Podczas diagnostyki badane są także parametry laboratoryjne. W szczególności następuje spadek stężenia testosteronu i czynnika krzepnięcia XII. Prowadzone są także studia instrumentalne. Zalecane jest prześwietlenie mostka, echokardiografia i EKG. Za pomocą tych metod wykrywa się anomalie narządów wewnętrznych. Lekarz może również umówić się na konsultację 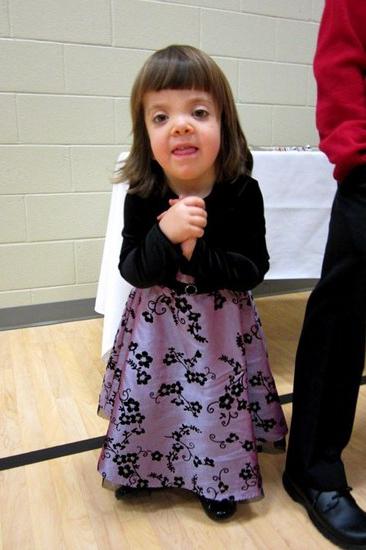
Środki terapeutyczne
Ze względu na genetyczny charakter zespołu leczenie ma na celu głównie eliminację objawów. W przypadku wnętrostwa zalecana jest operacja. Podczas tego procesu jądra przemieszczają się do moszny. Na tle spadku stężenia androgenów zaleca się terapię hormonalną. W przypadku niewydolności nerek zaleca się hemodializę. Za jego pomocą produkty przemiany materii są usuwane z organizmu poprzez pozanerkowe oczyszczanie krwi.
Zespół Noonana(J.A. Noonan, amerykański endokrynolog, ur. 1928) jest chorobą dziedziczną charakteryzującą się niskim wzrostem i nieprawidłowościami w rozwoju somatycznym. Najpełniej opisał ją J. Noonen w 1963 roku. Może mieć charakter sporadyczny lub rodzinny i dziedziczyć się w sposób autosomalny recesywny. Patologicznie zmienione geny powodujące N. s. są zlokalizowane w autosomach (patrz. Choroby dziedziczne ). Chromatyna płciowa i kariotyp (patrz. Chromosomy ) odpowiadają płci pacjenta. Choroba jest wykrywana zarówno u mężczyzn, jak iu kobiet.
Główne objawy kliniczne N. s. to niski wzrost, kończyny dolne, skrzydełkowate fałdy skórne na szyi, niska granica wzrostu włosów z tyłu głowy, charakterystyczne zmiany dermatoglificzne, hiperteloryzm, opadanie powiek, antymongoloidalny kształt oczu, deformacja lub niskie ustawienie płatków usznych , asymetria twarzy, szeroki grzbiet nosa, co sprawia, że pacjenci są do siebie podobni ( Ryż .). Charakteryzuje się krótką szyją, wrodzoną deformacją stawu łokciowego (łokieć koślawy), brachydaktylią, wysokim podniebieniem, patologiczną ugryzienie , płaskostopie, zmiany w kształcie i strukturze kręgów oraz inne anomalie, którymi się objawia Szereszewskiego – zespół Turnera . Często wykrywa się zwężenie płuc i ubytek przegrody międzykomorowej. Inne wrodzone wady rozwojowe układu sercowo-naczyniowego (patrz. Wrodzone wady serca ) są obserwowane rzadziej. Rozwój seksualny u N. s. waha się od normalnej do całkowitej dysgenezji gonad (patrz. Hermafrodytyzm ). Dziewczęta często doświadczają późnego początku miesiączki, podczas gdy chłopcy często doświadczają późnego początku miesiączki. wnętrostwo , hipogonadyzm . Badanie histologiczne materiału uzyskanego z biopsji jądra ujawnia zmniejszenie lub całkowity brak komórek rozrodczych i rozrost komórek Leydiga. Inne przejawy N. s. Czy ginekomastia i upośledzenie umysłowe. Surowica krwi pacjentów zawiera testosteron i estrogeny (patrz. Hormony płciowe ) i gonadotropiny (patrz. Hormony przysadkowe ) jest prawidłowe lub zmienia się w zależności od nasilenia dysgenezji gonad. Stężenie hormonu wzrostu we krwi jest w normie. Kariotyp 46XX lub 46XV odpowiada płci gonadalnej i paszportowej.
Rozpoznanie stawia się na podstawie charakterystycznych objawów klinicznych i danych laboratoryjnych. N. s. różnicowany najczęściej z zespołem Shereshevsky’ego-Turnera ( tabela .).
Leczenie objawów hipogonadyzmu polega na terapii zastępczej hormonami płciowymi. Stosowanie hormonu wzrostu jest nieskuteczne. Według wskazań wykonuje się chirurgiczną korekcję wad wrodzonych oraz leczenie zaburzeń psychicznych związanych z upośledzeniem umysłowym (patrz. Endokrynologiczne zaburzenia psychiczne ). Prognozy na całe życie są pomyślne. Szczególną uwagę należy zwrócić na zatrudnienie: wybór specjalności powinien przyczyniać się do adaptacji społecznej pacjentów, biorąc pod uwagę ich możliwości umysłowe i niepełnosprawność fizyczną.
Różnicowe objawy diagnostyczne zespołów Noonena i Shereshevsky’ego-Turnera
Znak kliniczny |
Zespół Noonana |
Zespół Szereszewskiego-Turnera |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Rozwój seksualny |
Zespół Noonana charakteryzuje się licznymi wadami wrodzonymi, typowymi dla pacjentów z zespołem Shereshevsky’ego-Turnera, z prawidłowym kariotypem. Czasami zespół ten utożsamia się z zespołem Bonneviego-Ullricha, który ma podobny obraz kliniczny. Jednak w przypadku zespołu Bonneviego-Ullricha nie ma wady serca, co bardzo często obserwuje się w przypadku zespołu Shereshevsky'ego-Turnera i Południe. U wszystkich pacjentów można wykryć mniej lub bardziej wyraźne objawy hipogonadyzmu, chociaż niektórzy autorzy uważają, że w przypadku zespołu Noonana u niektórych chłopców narządy płciowe rozwijają się normalnie. Etiologia. Zespół dziedziczy się w sposób autosomalny dominujący z niską penetracją genów. Rozkład zespołu jest dość duży (1:1000 – 1:5000), jednak nie ma jednoznacznego kryterium diagnostycznego w postaci nieprawidłowego kariotypu, jak u pacjentów z zespołem Shereshevsky’ego-Turnera. W rezultacie nie wszystkie przypadki choroby są wykrywane. Zależności podwzgórze-przysadka-gonady (patogeneza) w zespole Noonana praktycznie nie są badane. Istnieje tylko jedno doniesienie dotyczące cech układu rozrodczego pacjentki, na podstawie którego trudno dokonywać uogólnień. Zbadano poziom testosteronu i hormonów gonadotropowych we krwi oraz rezerwę czynnościową jąder u 8 chłopców z zespołem Noonana. Poziom testosteronu u wszystkich pacjentów był poniżej normy. Zmniejszone zostało także wydalanie 17-KS z moczem. Dane te wskazują na niedobór hormonów jąder w zespole Noonana. Stężenie testosteronu i hormonów gonadotropowych w surowicy krwi oraz wydalanie 17KC z moczem (M±m) u pacjentów z zespołem Noonana
Połączenie niewystarczającego wydzielania testosteronu i wysokiego poziomu LH u 4 pacjentów wskazuje na pierwotne uszkodzenie jąder, głównie komórek śródmiąższowych. Jednakże podawanie hCG przez 3 dni w dawce dziennej 1500 U/m2 spowodowało u tych chłopców znaczny wzrost poziomu testosteronu we krwi. Kauschunsky i in. (1977) podając GnRH pacjentowi z zespołem Noonana zaobserwowali wyraźny wzrost wydzielania gonadotropin, podobnie jak u pacjentów z pierwotnym hipogonadyzmem, i jednocześnie pozytywną reakcję jąder na hCG. Ograniczone obserwacje utrudniają interpretację danych. Można przypuszczać, że u pacjentów wrażliwość receptorów komórek Leydiga na endogenną gonadotropinę jest zmniejszona, ale rezerwy jąder zostają zachowane, co przekłada się na wzrost wydzielania testosteronu po stymulacji dużymi dawkami hCG. U pacjentów z niskim poziomem testosteronu i prawidłowym poziomem hormonów gonadotropowych we krwi patogeneza niewydolności jąder jest prawdopodobnie zbliżona do patogenezy hipogonadyzmu normogonadotropowego. Theintz i Savagl (1982) opisali pięciu chłopców w wieku 11,8–16,5 lat z zespołem Noonana. Wszystkie miały wyraźne objawy hipogonadyzmu i opóźniony rozwój płciowy. U 4 z nich rozwój seksualny powrócił do normy, u dwóch samoistnie, a u dwóch po leczeniu testosteronem. Tylko u jednego pacjenta leczenie hCG i testosteronem było nieskuteczne. Zatem u pacjentów z zespołem Noonana, pomimo powszechnego obrazu klinicznego, charakter hipogonadyzmu może być inny. Niewydolność ich układu rozrodczego może być konsekwencją pierwotnej wady gonad, dysfunkcji układu podwzgórzowo-przysadkowego i zmian wrażliwości jąder na stymulację gonadotropową. Obraz kliniczny przypomina klinikę zespołu Szereszewskiego-Turnera. Przedstawiamy wyciąg z historii choroby jednego z obserwowanych przez nas pacjentów. Serezha L., lat 8, została przyjęta na oddział endokrynologii dziecięcej szpitala im. Rauchfus, urodzony 5 marca 1982 r., z opóźnionym rozwojem fizycznym i umysłowym. Urodzony z drugiej ciąży, która wystąpiła z ciężką zatruciem. W 7-8 tygodniu ciąży matka przeszła ostrą infekcję wirusową dróg oddechowych. Pierwsza ciąża zakończyła się poronieniem. Matka cierpi na wrodzoną wadę. Chłopiec urodził się jako wcześniak (usunięty przez cesarskie cięcie). Masa ciała 3500 g, długość 51 cm, w okresie noworodkowym stan jest poważny. W 4 miesiącu życia zdiagnozowano wadę serca; chłopiec od pierwszego roku pozostawał w tyle w rozwoju fizycznym i umysłowym. Po urodzeniu wykryto brak jąder w mosznie. Przy przyjęciu długość ciała wynosiła 124 cm (poniżej średniej), masa ciała 22 kg. Budowa ciała jest nienormalna - szeroka, krótka szyja, asymetryczna klatka piersiowa, deformacja mostka w kształcie lejka; wysokie podniebienie, hiperteloryzm, deformacja uszu, wady palców, szeroko rozstawione sutki. Penis jest prawidłowo rozwinięty, moszna jest bardzo mała, jądra nie są wyczuwalne. Inteligencja jest umiarkowanie obniżona. Kariotyp 46XY. Rentgen czaszki bez cech. Wiek kostny odpowiada 7 latom. W surowicy krwi LH wynosi 4,33 mIU/ml, FSH 3,13 mIU/ml, testosteron 0,56 nmol/l (wszystkie wskaźniki są obniżone). Wydalanie 17KS wynosi 9,1 mmol/dzień. Test funkcjonalny po jednorazowym wstrzyknięciu hCG dał wynik negatywny – testosteron we krwi – 0,57 nmol/l, 17-KS – 8,87 mmol/dzień; Test 3-dniowy jest pozytywny - testosteron - 1,54 nmol/l, 17KS - 10,15 mmol/dzień. Na podstawie wywiadu, obrazu klinicznego i danych laboratoryjnych postawiono rozpoznanie zespołu Noonana. Dodatni wynik testu 3-dniowego pozwolił na leczenie hCG. Jeżeli nie ma efektu, wskazane jest chirurgiczne leczenie wnętrostwa. „Zaburzenia rozwoju płciowego u chłopców” Wygląd.
Formularze
Powoduje
DiagnostykaDiagnozę stawia się na podstawie:
Leczenie zespołu Noonana
Komplikacje i konsekwencje
Zapobieganie zespołowi Noonana
Podobne artykuły
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||