Sindromul Noonan și boli similare. Vezi ce este „Sindromul Noonan” în alte dicționare.
E.V. Tozliyan, endocrinolog pediatru, genetician, candidat la științe medicale, separat subdiviziune structurală„Institutul Clinic de Cercetare Științifică de Pediatrie” SBEI VPO Universitatea Națională de Cercetare Medicală din Rusia. N.I. Pirogov de la Ministerul Sănătății al Federației Ruse, Moscova Cuvinte cheie Cuvinte cheie: copii, sindrom Noonan, diagnostic.
Cuvinte cheie: copii, sindromul Noonan, diagnostice.
Articolul descrie sindromul Noonan (sindromul Ulrich-Noonan, sindromul terneroid cu un cariotip normal) - un rar patologie congenitală, moștenit în mod autosomal dominant, este caracter familial Cu toate acestea, apar și cazuri sporadice. Sindromul sugerează prezența unui fenotip caracteristic sindromului Shereshevsky-Turner la indivizii de sex feminin și masculin cu un cariotip normal. Prezentat observatie clinica. Complexitatea căutării diagnosticului diferențial, lipsa de conștientizare a clinicienilor despre acest sindromși importanța unei abordări interdisciplinare.
Fapte istorice
Pentru prima dată despre sindrom neobișnuit a menționat O. Kobylinski în 1883 (foto 1).
cel mai vechi cunoscut caz clinic Sindromul Noonan, descris în 1883 de O. Kobylinski
Boala a fost descrisă în 1963 de cardiologul american Jacqueline Noonan, care a raportat despre nouă pacienți cu stenoză valvulară. artera pulmonara, statură mică, hipertelorism, declin intelectual moderat, ptoză, criptorhidie și tulburări ale scheletului. Dr. Noonan, care a practicat ca cardiolog pediatru la Universitatea din Iowa, a observat că copiii cu tip rar boli de inimă - stenoza valvei pulmonare - au observat adesea anomalii fizice tipice sub formă de statură mică, gât pterigoid, ochi largi și urechi joase. Băieții și fetele au fost la fel de uimiți. Dr. John Opitz, fost student Noonan, a fost primul care a introdus termenul „sindrom Noonan” pentru a descrie starea copiilor care aveau semne similare cu cele descrise de Noonan. Ulterior, Noonan a scris articolul „Hipertelorism cu fenotipul Turner”, iar în 1971 numele „sindromul Noonan” a fost recunoscut oficial la simpozionul de boli cardiovasculare.
Etiologie și patogeneză
Sindromul Noonan este o tulburare autosomal dominantă cu expresivitate variabilă (Fig. 1). Gena sindromului Noonan este localizată la umăr lung cromozomul 12. Eterogenitatea genetică a sindromului nu poate fi exclusă. Au fost descrise forme sporadice și familiale ale sindromului cu o formă autosomal dominantă de moștenire. În cazurile familiale gena mutantă moștenit, de regulă, de la mamă, deoarece din cauza unor malformații severe sistemul genito-urinar bărbații cu această afecțiune sunt adesea infertili. Majoritatea cazurilor raportate sunt sporadice, cauzate de mutații de novo.
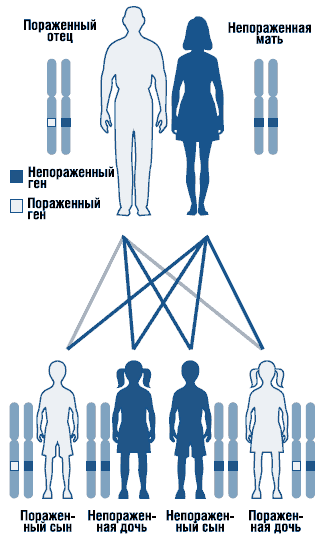
. Model de moștenire autozomal dominant
Combinațiile descrise ale sindromului Noonan cu neurofibromatoza de tip I în mai multe familii au sugerat o posibilă relație între doi loci independenți 17q11.2 ai cromozomului 17. Unii pacienți au microdeleții în locusul 22q11 al cromozomului 22; în aceste cazuri manifestari clinice Sindroamele Noonan sunt combinate cu hipotiroidismul timusului și cu sindromul DiGeorge. O serie de autori discută despre implicarea presupuselor gene ale limfogenezei în patogeneza sindromului datorită prezenței anomaliilor faciale și somatice similare cu sindromul Turner și a unei incidențe mari a patologiei. sistem limfatic.
Cel mai cauza comuna Sindromul Noonan este o mutație a genei PTPN11, care se găsește la aproximativ 50% dintre pacienți. Proteina codificată de gena PTPN11 aparține unei familii de molecule care reglează răspunsul celulelor eucariote la semnalele externe. Cel mai mare număr mutațiile din sindromul Noonan sunt localizate în exonii 3, 7 și 13 ai genei PTPN11, care codifică domeniile proteice responsabile de tranziția proteinei la starea activă.
Ideile posibile despre patogeneză sunt reprezentate de următoarele mecanisme:
Calea RAS-MAPK este o cale de transducție a semnalului foarte importantă prin care sunt liganzii extracelulari anumiți factori creșterea, citokinele și hormonii - stimulează proliferarea celulară, diferențierea, supraviețuirea și metabolismul (Fig. 2). După legarea ligandului, receptorii de pe suprafața celulei sunt fosforilați la locurile regiunii lor endoplasmatice. Această legare implică proteine adaptoare (de exemplu, GRB2) care formează un complex constitutiv cu factori de schimb de nucleotide de guanină (de exemplu, SOS) care convertesc RAS inactiv legat de GDP în forma sa activă legată de GTP. Proteinele RAS activate activează apoi cascada RAF-MEKERK printr-o serie de reacții de fosforilare. Ca rezultat, ERK activat intră în nucleu pentru a modifica transcripția genelor țintă și corectează activitatea țintelor endoplasmatice pentru a induce răspunsuri celulare adecvate pe termen scurt și lung la stimul. Toate genele implicate în sindromul Noonan codifică proteine parte integrantă a acestei căi și mutații cauzatoare de boli, de obicei amplifică semnalul care trece prin această cale.
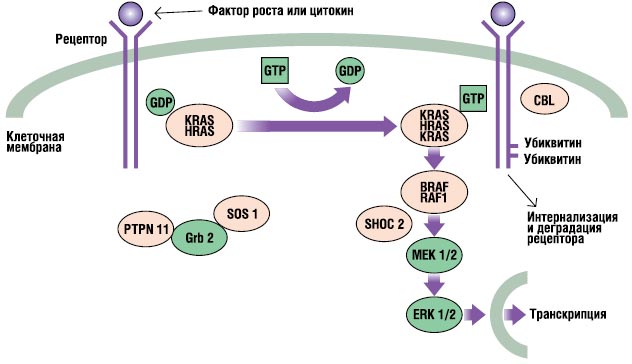
. Calea de semnalizare RAS-MAPK. Semnalele de creștere sunt transmise cu receptorii de factor de creștere activați către nucleu. Mutațiile în PTPN11, KRAS, SOS1, NRAS și RAF1 sunt asociate cu sindromul Noonan, iar mutațiile în SHOC2 și CBL sunt asociate cu un fenotip asemănător sindromului Noonan.
Caracteristicile clinice ale sindromului Noonan
Fenotipul pacienților cu sindromul Noonan seamănă cu sindromul Turner: gât scurt cu pliu pterigoidian sau creștere scăzută a părului, statură mică, hipertelorism al fisurilor palpebrale (foto 2). Microanomaliile faciale includ incizia antimongoloidă a fisurilor palpebrale, canthus lateral în jos, ptoza, epicantus, auricule joase, bucle pliate. auriculare, malocluzie, uvulă despicată palat moale, cerul gotic, micrognathia și microgenia. Toracele tiroidei se formează cu mameloane hipoplazice larg distanțate, sternul iese în partea superioară și se scufundă în partea inferioară. Aproximativ 20% dintre pacienți au o patologie moderată a scheletului. Cea mai frecventă deformare a pâlniei cufăr, cifoză, scolioză; mai rar - o scădere a numărului de vertebre cervicale și fuziunea acestora, asemănătoare cu anomalii în sindromul Klippel-Feil.
. Fenotipurile sindromului Noonan
Pacienții cu sindromul Noonan au, de obicei, părul blond, des, creț, cu creșterea neobișnuită a coroanei, adesea pete întunecate pe piele, hipertricoza, degenerarea plăcilor unghiale, anomalii în erupția și aranjarea dinților, tendința de a forma cicatrici cheloide, extensibilitatea pielii crescută. O treime dintre pacienți prezintă limfedem periferic, mai des limfedemul mâinilor și picioarelor se manifestă la copii vârstă fragedă. Un semn frecvent este patologia vederii (miopie, strabism, exoftalmie moderată etc.). Întârzierea creșterii apare la aproximativ 75% dintre pacienți, este mai pronunțată la băieți și este de obicei nesemnificativă. Întârzierea creșterii se manifestă în primii ani de viață, mai rar există un ușor deficit de creștere și greutate la naștere. Încă din primele luni de viață apare o scădere a poftei de mâncare. Vârsta osoasă este de obicei în urmă față de vârsta pașaportului.
O trăsătură caracteristică a sindromului este criptorhidia unilaterală sau bilaterală, care apare la 70-75% dintre pacienții de sex masculin; la pacienții adulți, azoospermia, oligospermia, modificări degenerative testicule. Cu toate acestea, pubertatea apare spontan, uneori cu o oarecare întârziere. La fete, există adesea o întârziere în formarea menstruației, uneori - încălcări ciclu menstrual. Fertilitatea poate fi normală la ambele sexe.
Retardarea mintală este detectată la mai mult de jumătate dintre pacienți, de obicei minor. Se remarcă adesea trăsături comportamentale, dezinhibarea, tulburarea de deficit de atenție. Vorbirea este de obicei mai dezvoltată decât altele sfere intelectuale. Gradul de scădere a inteligenței nu se corelează cu severitatea tulburărilor somatice [Marincheva G.S., 1988]. În cazuri izolate, malformații ale centralei sistem nervos(hidrocefalie, hernii spinale), infarcte tromboembolice ale creierului, posibil asociate cu hipoplazie vasculară.
vicii organe interne cu sindromul Noonan sunt destul de caracteristice. Cele mai tipice sunt anomaliile cardiovasculare: stenoza valvulară a arterei pulmonare (aproximativ 60% dintre pacienți), cardiomiopatie hipertropica(20–30%), anomalii structurale valva mitrala, defecte ale septului atrial, tetralogia Fallot; coarctația aortei a fost descrisă numai la pacienții de sex masculin.
La o treime dintre pacienți se înregistrează malformații ale sistemului urinar (hipoplazie a rinichilor, dublarea pelvisului, hidronefroză, megaureter etc.).
Destul de des, cu sindromul Noonan, se observă sângerare crescută, în special cu interventii chirurgicale V cavitatea bucalăși nazofaringe. Se constată diverse defecte de coagulare: insuficiență a sistemului trombocitar, scăderea nivelului factorilor de coagulare, în special XI și XII, creșterea timpului de tromboplastină. Există rapoarte privind o combinație a sindromului Noonan cu leucemie și rabdomiosarcom, care poate indica o ușoară creștere a riscului de malignitate la acești pacienți.
Tabelul 1 prezintă caracteristicile fenotipului în sindromul Noonan, care se modifică odată cu vârsta pacientului. Tabelul 2 arată corelația dintre fenotip și genotip în sindromul Noonan.
tabelul 1. Trăsături faciale tipice la pacienții cu sindrom Noonan în funcție de vârstă
| Frunte, față, păr | Ochi | Urechi | Nas | Gură | Gât | |
| Nou nascut* | Frunte înaltă, linie joasă a părului în regiunea occipitală | Hipertelorism, înclinat descendent fisuri palpebrale, pliu epicantal | – | Rădăcină scurtă și largă îngroșată, vârful răsturnat | Filtru adânc îngropat, vârfuri înalte și largi ale marginii vermilion a buzelor, micrognatie | Exces de piele pe spatele capului |
| Sân (2-12 luni) | Cap mare, frunte înaltă și proeminentă | Hipertelorism, ptoză sau pleoape groase căzute | – | Rădăcină adâncită scurtă și largă | – | – |
| Copil (1-12 ani) | Trăsături aspre, față lungă | – | – | – | – | – |
| Adolescent (12-18 ani) | Față miopată | – | – | Podul este înalt și subțire | – | Formare evidentă a pliului gâtului |
| Adult (>18 ani) | Trăsăturile faciale distinctive sunt rafinate, pielea pare subțire și translucidă | – | – | Pliul nazolabial proeminent | – | – |
| Toate varstele | – | Iriși albaștri și verzi, sprâncene în formă de romb | Urechi joase, rotite în spate, cu pliuri groase | – | – | – |
masa 2. Corelații între genotip și fenotip în sindromul Noonan*
| Sistemul cardiovascular | Înălţime | Dezvoltare | Pielea și părul | Alte | |
| PTPN11 (aproximativ 50%) | Stenoza unui trunchi pulmonar este mai exprimată; mai puțin – cardiomiopatie hipertrofică și defect septal atrial | Creștere mai scăzută; concentrație mai mică de IGF1 | Pacienții cu N308D și N308S au un declin ușor sau o inteligență normală | – | Diateză hemoragică mai pronunțată și leucemie mielomonocitară juvenilă |
| SOS1 (aproximativ 10%) | Mai puțin defect septal atrial | Creștere mai mare | Mai puțin declin al inteligenței, întârzierea dezvoltării vorbirii | Similar cu sindromul facial cardiocutanat | – |
| RAF1 (aproximativ 10%) | Cardiomiopatie hipertrofică mai severă | – | – | Mai mult semne de nastere, lentigo, pete de cafea cu lapte | – |
| KRAS (<2%) | – | – | Întârziere cognitivă mai severă | Similar cu sindromul cardio-cutanat-facial | – |
| NRAS (<1%) | – | – | – | – | – |
Date din studii de laborator și funcționale
Nu există markeri biochimici specifici pentru diagnosticul sindromului Noonan. La unii pacienți, o scădere a secreției spontane nocturne a hormonului de creștere cu un răspuns normal la testele de stimulare farmacologică (clofelină și arginină), o scădere a nivelului de somatomedin-C și o scădere a răspunsului somatomedinelor la introducerea hormonului de creștere. sunt detectate.
Criterii de diagnostic
Diagnosticul „sindromului Noonan” se face pe baza semnelor clinice, în unele cazuri diagnosticul este confirmat de rezultatele unui studiu genetic molecular. Criteriile de diagnosticare a sindromului includ prezența unei fețe caracteristice (cu un cariotip normal) în combinație cu una dintre următoarele caracteristici: boli de inimă, statură mică sau criptorhidie (la băieți), pubertate întârziată (la fete). Pentru a detecta patologia cardiovasculară, este necesar să se efectueze o examinare cu ultrasunete a inimii cu o determinare dinamică a dimensiunii cavităților și a pereților ventriculilor. Diagnosticul prenatal al bolii este posibil cu ajutorul monitorizării cu ultrasunete, ceea ce face posibilă detectarea defectelor cardiace și a anomaliilor în structura gâtului.
Diagnostic diferentiat
La fete, diagnosticul diferenţial se face în primul rând cu sindromul Turner; Diagnosticul poate fi clarificat prin examen citogenetic. Semnele fenotipice ale sindromului Noonan se găsesc într-o serie de alte boli: sindromul Williams, sindromul LEOPARD, Dubovitz, sindromul cardiofacio-cutanat, Cornelia de Lange, Cohen, Rubinstein-Taybi, etc. Identificarea precisă a acestor boli va fi posibilă numai atunci când se efectuează studii genetice moleculare ale fiecărui sindrom cu material clinic semnificativ care este în prezent dezvoltat activ.
Tratament
Tratamentul pacienților cu sindrom Noonan are ca scop eliminarea defectelor sistemului cardiovascular, normalizarea funcțiilor mentale, stimularea creșterii și dezvoltării sexuale. Pentru tratamentul pacienților cu displazie a valvelor arterei pulmonare, printre alte metode, se utilizează cu succes valvuloplastia cu balon. Pentru a stimula dezvoltarea psihică se folosesc agenți nootropi și vasculari. Medicamentele care vizează stimularea dezvoltării sexuale sunt indicate în principal pacienților cu criptorhidie. Preparatele de gonadotropină corionică sunt utilizate în doze de vârstă. La o vârstă mai înaintată - în prezența hipogonadismului - preparate cu testosteron. În ultimii ani, forme recombinate ale hormonului uman de creștere au fost folosite în tratamentul pacienților cu sindrom Noonan. Datele clinice sunt confirmate de o creștere a nivelului de somatomedină-C și a proteinei de legare specifică în timpul terapiei. Înălțimea finală a pacienților care primesc terapie cu hormon de creștere pe termen lung, în unele cazuri, depășește înălțimea medie a membrilor familiei.
Prognoza căci viața este determinată de severitatea patologiei cardiovasculare.
Prevenirea boala se bazează pe datele consilierii genetice medicale.
Consiliere genetică medicală
În consilierea medicală genetică, trebuie să se procedeze de la tipul de moștenire autozomal dominant și un risc ridicat (50%) de reapariție a bolii în familia cu forme moștenite. Pentru a identifica natura tipului de moștenire, este necesar să se efectueze o examinare amănunțită a părinților, deoarece sindromul se poate manifesta cu simptome clinice minime. În prezent, diagnosticul genetic molecular al bolii a fost dezvoltat și este îmbunătățit prin tiparea mutațiilor în genele: PTPN11, SOS1, RAF1, KRAS, NRAS, etc. Se dezvoltă metode de diagnostic prenatal al bolii.
Observație clinică
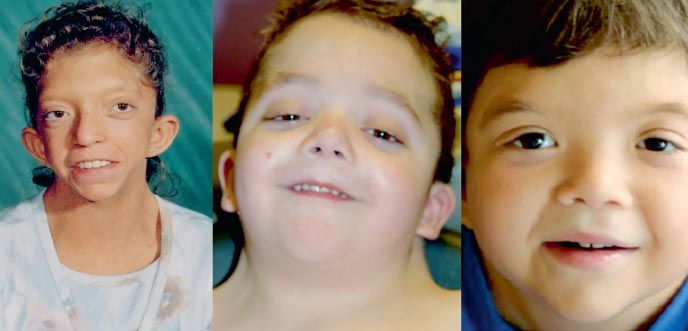
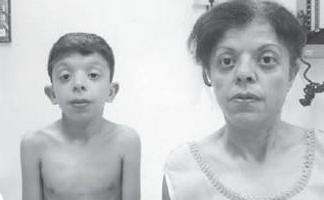
Băiatul G., în vârstă de 9 ani (foto 3), a fost observat la locul de reședință de un genetician cu diagnostic de patologie cromozomială?, sindrom Williams (fenotip deosebit, îngroșarea cuspidelor valvei mitrale, hipercalcemie o dată la 3 ani) ?.
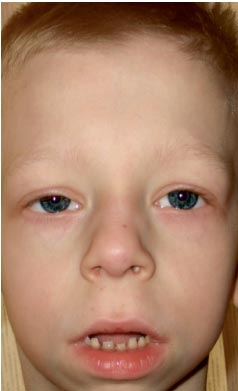
. Particularități ale fenotipului unui copil cu sindrom Noonan (un schelet facial alungit cu „obraji dolofani”, un gât scurt, pliuri pterigoide pe gât, un nas scurtat cu nările deschise înainte, buzele umflate, o bărbie înclinată, un anti-mongoloid incizia fisurilor palpebrale, malocluzie, macrostomie)
Reclamații pe memorie redusă, oboseală, rate de creștere reduse.
Istorie de familie : parintii sunt rusi dupa nationalitate, nu sunt rude de sange si nu prezinta riscuri profesionale, sanatosi. Înălțimea tatălui este de 192 cm, înălțimea mamei este de 172 cm. În pedigree-ul cazurilor de boli mintale, epilepsie, întârzieri de dezvoltare nu au fost notate.
Istoria vieții și a bolii : un băiat din a 2-a sarcină (1-a sarcină - m / a), care a continuat cu amenințarea întreruperii pe tot parcursul, însoțită de polihidramnios. Nașterea a fost prima, la timp, rapidă, greutatea la naștere - 3400 g, lungime - 50 cm. A țipat imediat, scor Apgar - 7/9 puncte. La naștere, medicul neonatolog a atras atenția asupra fenotipului neobișnuit al copilului, a recomandat studiul cariotipului, rezultatul este 46, XY (cariotip masculin normal). S-a suspectat hipotiroidism congenital, s-a efectuat un studiu de profil tiroidian, rezultatul a fost o stare tiroidiană normală. Mai mult, copilul a fost observat de un genetician cu un presupus diagnostic de „sindrom Williams”. Perioada postnatală timpurie - fără caracteristici. Dezvoltarea motorie în funcție de vârstă, primele cuvinte - după an, vorbire frazală - la 2 ani și 3 luni.
La vârsta de 8 ani, a fost consultat de un endocrinolog despre ritmul de creștere redus, oboseală și memoria redusă. O examinare cu raze X a mâinilor a evidențiat un decalaj moderat al vârstei osoase (BC) față de cea a pașaportului (BC corespundea la 6 ani). Studiul profilului tiroidian a relevat o creștere moderată a hormonului de stimulare a tiroidei cu un nivel normal de T4 liber și alți indicatori; Ecografia glandei tiroide - fără patologie. A fost prescrisă terapia hormonală, urmată de observație dinamică.
Ținând cont de incertitudinea diagnosticului la locul de reședință, geneticianul a îndrumat copilul către Centrul Regional Consultativ și Diagnostic pentru Copii din Moscova pentru a clarifica diagnosticul.
Date obiective ale cercetării:
Înălțime - 126 cm, greutate - 21 kg.
Dezvoltarea fizică este sub medie, armonioasă. Sds de creștere corespunde cu -1 (normal -2 + 2). Caracteristici ale fenotipului (foto 3): schelet facial alungit cu „obraji dolofani”, gat scurt, pliuri pterigoide pe gat, crestere redusa a parului pe gat, nas scurt cu nările deschise înainte, buze umflate, bărbie înclinată, anti-mongoloid incizie a fisurilor palpebrale, malocluzie, macrostomie, hipertelorism mamelon, asimetrie a toracelui, sindactilie cutanată incompletă a degetelor 2 sau 3 la picioare, hipermobilitate pronunțată a articulațiilor interfalangiene, unghii fragile, uscate. Pe organele interne - fără caracteristici. Dezvoltarea sexuală – Tanner I (care corespunde perioadei prepuberale).
Date din studii de laborator și funcționale:
Analiza clinică a sângelui și urinei este norma.
Analiza biochimică a sângelui - indicatori în intervalul normal.
Profilul tiroidian (TSH) - 7,5 μUI / ml (normal - 0,4-4,0), alți indicatori sunt normali.
Hormon somatotrop (STH) - 7 ng / ml (normal - 7-10), somatomedin-C - 250 ng / ml (normal - 88-360).
Ecografia glandei tiroide - fără patologie.
Ecografia organelor interne - fără caracteristici.
ECG - tahicardie sinusală, poziția normală a axei electrice a inimii.
EchoCG - MVP de gradul I cu regurgitare minimă, îngroșare mixomatoasă a cuspidelor valvei mitrale, o coardă suplimentară în cavitatea ventriculului stâng.
R-grafia coloanei vertebrale - scolioza dreapta a coloanei vertebrale toracice, gradul I.
R-grafie a mâinilor cu capturarea antebrațelor - vârsta osoasă 7–8 ani.
Modelele EEG ale activității epileptice nu au fost înregistrate.
RMN al creierului - fără modificări patologice.
Audiograma - fără patologie.
Diagnosticarea ADN: studiu genetic molecular - nu au fost detectate deleții ale locilor studiati din regiunea critică a cromozomului 7; Mutația Gly434Ary (1230G>A) a fost găsită în al 11-lea exon al genei SOS1 (analiza genei PTPN11 - nu au fost găsite mutații), ceea ce este tipic pentru sindromul Noonan.
Consultanță de specialitate:
Endocrinolog- hipotiroidism subclinic, compensare medicamentoasă incompletă.
optometrist- astigmatism.
Neurolog- distonie vegetativă. reacții nevrotice.
Cardiolog- cardiopatie functionala.
Chirurg ortoped- încălcarea posturii. Deformare toracică.
Genetician sindromul Noonan.
Luând în considerare fenotipul copilului, datele istorice, rezultatele studiilor suplimentare, a fost pus diagnosticul sindromului Noonan, care a fost confirmat de rezultatul unui studiu genetic molecular.
Astfel, observația clinică prezentată demonstrează complexitatea căutării diagnosticului diferențial, necesitatea de a integra semnele individuale în fenotipul general al unei anumite afecțiuni patologice pentru diagnosticarea în timp util a anumitor forme de boli ereditare și importanța metodelor genetice moleculare pentru a clarifica diagnostic. Diagnosticul în timp util, clarificarea genezei fiecărui sindrom sunt deosebit de importante, deoarece vă permit să găsiți cea mai bună abordare a tratamentului acestor afecțiuni, prevenirea posibilelor complicații (până la handicapul copilului); prevenirea recidivei bolilor ereditare în familiile afectate (consiliere genetică medicală). Acest lucru dictează necesitatea medicilor de diferite specialități de a naviga în mod clar în fluxul patologiei ereditare.
Bibliografie:
- Baird P., Sindromul De Jong B. Noonan (fenotipul XX și XY Turner) în trei generații ale unei familii // J. Pediatr., 1972, voi. 80, p. 110–114.
- Hasegawa T., Ogata T. şi colab. Coarctarea aortei și a hupoplaziei renale la un băiat cu anomalii de suprafață Turner/Noonan și un cariotip 46, XY: un model clinic pentru posibila afectare a unei gene limfogenice presupuse pentru stigmate somatice Turner // Hum. Genet., 1996, voi. 97, p. 564–567.
- Fedotova T.V., Kadnikova V.A. et al. Analiza clinico-molecular-genetică a sindromului Noonan. Materiale ale celui de-al VI-lea Congres al Societății Ruse de Genetică Medicală. Genetică medicală, Supliment la nr. 5, 2010, p.184.
- Ward K.A., Moss C., McKeown C. Sindromul cardio-facio-cutanat: o manifestare a sindromului Noonan? // Br. J. Dermatol., 1994, voi. 131, p. 270–274.
- Municchi G., Pasquino A.M. et al. Tratamentul cu hormon de creștere în sindromul Noonan: raportul a patru cazuri care au atins înălțimea finală // Horm. Res., 1995, voi. 44, p. 164–167.
Există diferite patologii congenitale la copii. Unele dintre ele sunt considerate destul de comune. Dar există și boli rare. Prevalența lor este scăzută. Cu toate acestea, multe au consecințe destul de grave. Una dintre aceste patologii este sindromul Noonan. Apoi, luați în considerare mai detaliat. Articolul va descrie cum și de ce apare sindromul Noonan. În text va fi prezentată și o fotografie a bolii.
Informații generale
Sindromul Noonan este o patologie ereditară. Gena mutantă PTPN11 de la părinții care o poartă este transmisă descendenților. De regulă, bărbații sunt infertili. Prin urmare, gena este transmisă prin linia maternă. Natura familială a acestei boli este de obicei remarcată. Cazuri rare care nu au legătură cu ereditatea, totuși, ele sunt înregistrate și în practică.
Referință istorică
La un moment dat, Jacqueline Noonan, care a profesat ca cardiolog pediatru, în timp ce lucra la o clinică de la Universitatea din Iowa, a observat că un anumit grup de copii, care includea atât băieți, cât și fete care aveau stenoză pulmonară, diferă adesea în ceea ce privește statura mică, cu ochi largi, gât palmeat. , localizate auricule joase și ptoză. După ce a examinat combinația de manifestări ale bolilor de inimă cu alte anomalii de dezvoltare la 833 de pacienți, medicul a scris un articol în 1962. În munca sa, ea a descris nouă cazuri în care trăsăturile faciale caracteristice și statura mică au fost observate pe fundalul tipului congenital. Boala a fost găsită atât la bărbați, cât și la femei. 
Sindromul Noonan: simptome
Există o serie de trăsături caracteristice care disting patologia de altele. Acestea includ, în special:
- Crestere inceata. Pentru femei - 1,53, pentru bărbați - 1,63 m. În momentul nașterii, atât lungimea corpului, cât și greutatea copilului sunt în limitele normale. Întârzierea creșterii începe la vârsta de 2-3 ani.
- Se schimbă chipul. Pacienții diagnosticați cu sindromul Noonan au forme larg distanțate. Există un pliu de piele în colțul interior. Există și ptoză (pleoape căzute) din cauza funcției musculare slăbite sau din cauza orbitelor mici. Aproximativ două treimi din toți copiii cu patologie au strabism.Când ptoza este limitată și se dezvoltă strabismul.
- Încălcări ale structurii maxilarelor. Cel de sus este subdezvoltat, există un palat înalt arcuit. La ambele maxilare, poziția dinților este incorectă.
- Aterizare scăzută sau deformare a auricularelor. Ca urmare a acestei anomalii, auzul este afectat.
- Gât scurt și larg.
- Protejați pieptul cu sfarcurile distanțate larg.
- Deformarea articulației cotului (congenitală).
- Picioare plate.
- Degete scurte.
Malformații ale organelor interne
Sindromul Noonan este cel mai adesea însoțit de tulburări în activitatea sistemului cardiovascular. Pe fondul patologiei, se detectează o îngustare (stenoză) a trunchiului pulmonar, precum și un defect al septului interventricular. Al doilea loc este ocupat de anomalii ale sistemului genito-urinar. Deci, din partea rinichilor, sunt relevate defecte precum hipoplazia (lipsa dezvoltării țesuturilor) sau absența unui singur rinichi. În ceea ce privește pubertatea, aceasta variază de la normal la complet defectuos. Fetele au adesea un debut tardiv al menstruației, băieții au criptorhidie sau testiculele sunt complet absente. Există, de asemenea, încălcări ale spermatogenezei. În unele cazuri, spermatozoizii sunt complet absenți. Pe fondul operațiilor cu sindromul Noonan, se observă o creștere a sângerării. Unii pacienți pot prezenta, de asemenea, anomalii ușoare. 
Forme
Există două tipuri de patologie:
- forma familiei. Diferă prin natura ereditară a tipului autosomal dominant. La purtătorii genei mutante, descendenții apar cu patologie.
- formă sporadică. În acest caz, mutația apare de la caz la caz. În acest caz, factorul ereditar nu a fost identificat.
Cauze
Cel mai frecvent factor care provoacă patologia este o mutație a genei PTPN11. Această cauză este detectată la 50% dintre pacienți. Cu toate acestea, la un anumit procent din persoanele cu acest sindrom, factorul genetic este neclar. Moștenirea patologiei are loc într-o formă autosomal dominantă. Sindromul poate fi declanșat de o nouă mutație. De aici rezultă că părinții cu o astfel de genă nu au șanse mari să aibă un alt copil.
Diagnostic
Este instalat în conformitate cu caracteristicile externe caracteristice (sunt descrise mai sus). În timpul diagnosticului sunt examinați și indicatorii de laborator. În special, există o scădere a concentrației de testosteron, factor de coagulare XII. De asemenea, sunt efectuate studii instrumentale. Se prescrie o radiografie a sternului, ecocardiografie, ECG. Cu ajutorul acestor metode sunt detectate anomalii ale organelor interne. De asemenea, medicul poate programa o consultație 
Activitati terapeutice
Datorită stării genetice a sindromului, tratamentul vizează în principal eliminarea simptomelor. Cu criptorhidie, este prescrisă o operație. În timpul acesteia, testiculele se deplasează în scrot. Pe fondul scăderii concentrației de androgeni, se recomandă terapia hormonală. În prezența insuficienței renale, este prescrisă hemodializă. Cu ajutorul acestuia, produsele proceselor metabolice sunt îndepărtate din organism prin purificarea sângelui extrarenal.
sindromul Noonan(J.A. Noonan, medic endocrinolog american, născut în 1928) este o boală ereditară caracterizată prin statură mică și anomalii ale dezvoltării somatice. J. Nunen este descris cel mai complet în 1963. Poate fi sporadic sau familial, moștenit într-o manieră autosomal recesiv. Genele modificate patologic care cauzează N. de pagină sunt localizate în autozomi (vezi. boli ereditare ). Cromatina sexuală și cariotip Cromozomii ) se potrivesc cu sexul pacientului. Boala apare atât la bărbați, cât și la femei.
Principalele manifestări clinice ale N. cu. statură mică, extremități inferioare, pliuri pterigoide ale pielii pe gât, linia scăzută a părului în partea din spate a capului, modificări dermatoglifice caracteristice, hipertelorism, ptoză, incizie antimongoloidă a ochilor, deformare sau potrivire scăzută a marginilor urechii, asimetrie a urechii. fata, o punte larga a nasului, care face ca pacientii sa fie asemanatori intre ei prieteni ( orez .). Caracterizat prin gât scurt, deformare congenitală a articulației cotului (cubitus valgus), brahidactilie, palat înalt, patologic mușcă , picioare plate, modificări ale formei și structurii vertebrelor și alte anomalii observate în timpul Sindromul Shereshevsky-Turner . Adesea găsită stenoză a trunchiului pulmonar, defect septal ventricular. Alte malformații congenitale ale sistemului cardiovascular (vezi. Malformații cardiace congenitale ) sunt mai puțin frecvente. Dezvoltarea sexuală la N. pag. variază de la normal la disgeneza gonadală completă (vezi Hermafroditismul ). Fetele au adesea un debut tardiv al menstruației, băieții - criptorhidie , hipogonadism . Examenul histologic al materialului obținut din biopsia testiculară relevă scăderea sau absența completă a celulelor germinale și hiperplazia celulelor Leydig. Alte manifestări ale N. cu. sunt ginecomastie si retard mintal. În serul sanguin al pacienților, conținutul de testosteron, estrogeni (vezi. hormoni sexuali ) și gonadotropine (vezi hormoni pituitari ) este normală sau modificată în funcție de severitatea disgenezei gonadale. Concentrația hormonului de creștere în sânge este normală. Cariotipul 46XX sau 46XV corespunde sexului gonadal și pașaportului.
Diagnosticul se stabilește pe baza semnelor clinice caracteristice și a datelor de laborator. N. s. cel mai adesea diferențiat cu sindromul Shereshevsky-Turner ( masa .).
Tratamentul în prezența semnelor de hipogonadism constă în terapia de substituție cu preparate cu hormoni sexuali. Utilizarea hormonului de creștere este ineficientă. Conform indicațiilor, corectarea chirurgicală a malformațiilor congenitale, tratamentul tulburărilor mintale asociate cu retardul mintal (vezi. Tulburări mentale endocrine ). Prognosticul pentru viață este favorabil. O atenție deosebită trebuie acordată ocupării forței de muncă: alegerea specialității ar trebui să contribuie la adaptarea socială a pacienților, ținând cont de abilitățile lor mentale și de dizabilitățile fizice.
Caracteristicile diagnostice diferențiale ale sindroamelor Noonan și Shereshevsky-Turner
semn clinic |
sindromul Noonan |
Sindromul Shereshevsky-Turner |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
dezvoltarea sexuală |
Sindromul Noonan se caracterizează prin multiple malformații congenitale tipice pacienților cu sindrom Shereshevsky-Turner, cu un cariotip normal. Uneori, sindromul este identificat cu sindromul Bonnevie-Ulrich, care are un tablou clinic similar. Cu toate acestea, cu sindromul Bonnevie-Ulrich, nu există nicio boală de inimă, care este foarte des observată în sindromul Shereshevsky-Turner și Noonan. La toți pacienții pot fi depistate simptome mai mult sau mai puțin pronunțate de hipogonadism, deși unii autori consideră că, în cazul sindromului Noonan, organele genitale se dezvoltă normal la unii băieți. Etiologie. Sindromul este moștenit într-o manieră autozomal dominantă cu penetranță scăzută a genelor. Distribuția sindromului este destul de mare (1:1000 - 1:5000), dar nu există un criteriu de diagnostic clar sub forma unui cariotip anormal, ca la pacienții cu sindrom Shereshevsky-Turner. În acest sens, nu sunt detectate toate cazurile de boală. Relațiile hipotalamo-hipofizo-gonadale (patogeneza) în sindromul Noonan nu sunt practic studiate. Există un singur raport despre caracteristicile sistemului reproducător la un pacient, pe baza căruia este dificil să se facă generalizări. Am investigat conținutul de testosteron și hormoni gonadotropi din sânge, precum și rezervele funcționale ale testiculelor la 8 băieți cu sindrom Noonan. Nivelurile de testosteron la toți pacienții au fost sub normal. Excreția urinară de 17-KS a fost de asemenea redusă. Aceste descoperiri indică o deficiență hormonală testiculară în sindromul Noonan. Nivelurile serice de testosteron și hormoni gonadotropi și excreția urinară de 17KS (M±m) la pacienții cu sindrom Noonan
Combinația dintre secreția insuficientă de testosteron și niveluri ridicate de LH la 4 pacienți indică o leziune primară a testiculelor, în principal celule interstițiale. Cu toate acestea, administrarea de CG timp de 3 zile la o doză zilnică de 1500 UI/m2 a dus la o creștere semnificativă a nivelului de testosteron din sânge la acești băieți. Kauschunsky și colab. (1977) la administrarea GnRH unui pacient cu sindrom Noonan, au observat o creștere pronunțată a secreției de gonadotropine, ca la pacienții cu hipogonadism primar, și, în același timp, o reacție pozitivă a testiculelor la CG. Numărul limitat de observații face dificilă interpretarea datelor. Se poate presupune că la pacienți sensibilitatea receptorilor celulelor Leydig la gonadotropina endogene este redusă, dar rezervele testiculelor sunt păstrate, ceea ce se exprimă într-o creștere a secreției de testosteron după stimularea cu doze mari de hCG. La pacienții cu niveluri scăzute de testosteron și niveluri normale de hormoni gonadotropi în sânge, patogenia insuficienței testiculare este probabil apropiată de patogeneza hipogonadismului normogonadotrop. Theintz, Savagl (1982) a descris 5 băieți cu vârsta cuprinsă între 11,8 și 16,5 ani cu sindromul Noonan. Toți aveau semne clare de hipogonadism și au rămas în urmă în dezvoltarea sexuală. În 4 dintre ele, apoi dezvoltarea sexuală a revenit la normal, iar în două spontan, iar în două - după tratament cu testosteron. Doar la un pacient, tratamentul cu CG și testosteron a fost ineficient. Astfel, la pacienții cu sindrom Noonan, în ciuda caracterului comun al tabloului clinic, natura hipogonadismului poate fi diferită. Insuficiența sistemului reproducător în ele poate fi rezultatul unui defect primar al gonadelor, disfuncție a sistemului hipotalamo-hipofizar și modificări ale sensibilității testiculelor la stimularea gonadotropă. Tabloul clinic seamănă cu clinica sindromului Shereshevsky-Turner. Iată un extras din istoricul unuia dintre pacienții pe care i-am observat. Serezha L., în vârstă de 8 ani, a fost internată în secția de endocrinologie pediatrică a spitalului. Rauhfus pe 03/05/82 cu o întârziere în dezvoltarea fizică și mentală. S-a născut din a doua sarcină, care a continuat cu toxicoză severă. În a 7-a - a 8-a săptămână de sarcină, mama a suferit o infecție virală respiratorie acută. Prima sarcină s-a încheiat cu un avort spontan. Mama suferă de boli congenitale Băiatul s-a născut prematur (extras prin cezariană). Greutatea corporală 3500 g, lungime 51 cm.În perioada neonatală, starea este severă. La vârsta de 4 luni a fost diagnosticată o boală de inimă; baiatul a ramas in urma in dezvoltarea fizica si psihica inca din primul an. Absența testiculelor în scrot a fost detectată la naștere. La internare, lungimea corpului 124 cm (sub medie), greutate 22 kg. Fizicul este incorect - un gât larg scurt, un piept asimetric, o deformare în formă de pâlnie a sternului; palat înalt, hipertelorism, deformare a urechii, defecte ale degetelor, mameloane distanțate larg. Penisul este dezvoltat satisfăcător, scrotul este foarte mic, testiculele nu sunt palpabile. Inteligența este moderat redusă. Cariotip 46XY. Radiografia craniului fără trăsături. Vârsta osoasă corespunde la 7 ani. În serul de sânge LH 4,33 mIU / ml, FSH 3,13 mIU / ml, testosteron 0,56 nmol / l (toți indicatorii sunt redusi). Excreție 17KS 9,1 mmol/zi. Testul funcțional cu o singură injecție de hCG este negativ - testosteron din sânge - 0,57 nmol/l, 17-KS - 8,87 mmol/zi; Testul de 3 zile este pozitiv - testosteron - 1,54 nmol / l, 17KS - 10,15 mmol / zi. Pe baza anamnezei, a prezentării clinice și a datelor de laborator, a fost pus un diagnostic al sindromului Noonan. Un test pozitiv de 3 zile a permis tratamentul CG. În absența efectului, este indicat tratamentul chirurgical al criptorhidiei. „Tulburări ale dezvoltării sexuale la băieți”, Aspect.
Forme
Cauze
DiagnosticareDiagnosticul se stabilește pe baza:
Tratamentul sindromului Noonan
Complicații și consecințe
Prevenirea sindromului Noonan
Articole similare
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||