Noonanov sindrom i slične bolesti. Pogledajte šta je "Noonan sindrom" u drugim rječnicima.
E.V. Tozliyan, pedijatar endokrinolog, genetičar, kandidat medicinskih nauka, Separat strukturna podjela"Naučno-istraživački klinički institut za pedijatriju" SBEI VPO Ruski nacionalni istraživački medicinski univerzitet. N.I. Pirogova Ministarstva zdravlja Ruske Federacije, Moskva Ključne riječi Ključne riječi: djeca, Noonan sindrom, dijagnostika.
ključne reči: djeca, Noonan sindrom, dijagnostika.
U članku se opisuje Noonan sindrom (Ulrich-Noonan sindrom, terneroidni sindrom s normalnim kariotipom) - rijedak kongenitalna patologija, naslijeđen na autosomno dominantan način, je porodični karakter Međutim, javljaju se i sporadični slučajevi. Sindrom ukazuje na prisustvo fenotipa karakterističnog za Shereshevsky-Turnerov sindrom kod ženskih i muških osoba s normalnim kariotipom. Presented kliničko posmatranje. Složenost diferencijalne dijagnostičke pretrage, nedostatak svijesti kliničara o ovaj sindrom i važnost interdisciplinarnog pristupa.
Istorijske činjenice
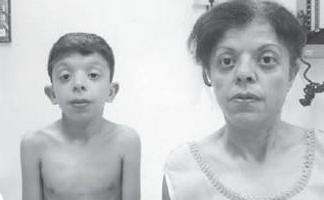
Po prvi put o neobičan sindrom pominje O. Kobylinski 1883. (slika 1).
najstariji poznati klinički slučaj Noonanov sindrom, koji je 1883. opisao O. Kobylinski
Bolest je 1963. godine opisala američka kardiologinja Jacqueline Noonan, koja je izvijestila o devet pacijenata sa stenozom zalistaka. plućna arterija, nizak rast, hipertelorizam, umjereni intelektualni pad, ptoza, kriptorhizam i skeletni poremećaji. Dr Noonan, koji je praktikovao kao pedijatrijski kardiolog na Univerzitetu Iowa, primijetio je da djeca sa rijedak tip srčana bolest - stenoza plućne valvule - često se uočavaju tipične fizičke anomalije u vidu niskog rasta, pterigoidnog vrata, široko postavljenih očiju i nisko postavljenih ušiju. Dečaci i devojčice su bili podjednako zadivljeni. dr John Opitz, bivši student Noonan, je bio prvi koji je uveo termin "Noonan sindrom" kako bi opisao stanje djece koja su imala znakove slične onima koje je opisao Noonan. Kasnije je Noonan napisao članak "Hipertelorizam sa Turnerovim fenotipom", a 1971. godine na simpozijumu kardiovaskularnih bolesti zvanično je priznat naziv "Noonanov sindrom".
Etiologija i patogeneza
Noonan sindrom je autosomno dominantni poremećaj sa promjenjivom ekspresivnošću (slika 1). Gen za Noonan sindrom je lokaliziran na dugo rame hromozom 12. Ne može se isključiti genetska heterogenost sindroma. Opisani su sporadični i porodični oblici sindroma sa autosomno dominantnim oblikom nasljeđivanja. U porodičnim slučajevima mutantni gen naslijeđen, po pravilu, od majke, jer zbog teških malformacija genitourinarnog sistema muškarci sa ovim stanjem često su neplodni. Većina prijavljenih slučajeva je sporadična, uzrokovana de novo mutacijama.
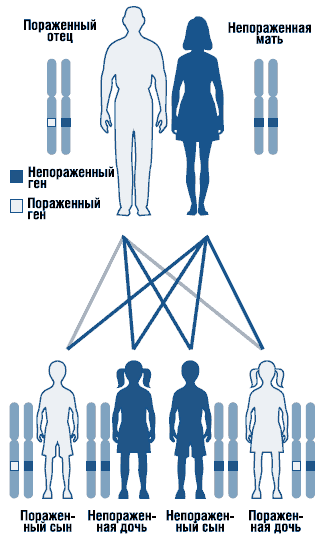
. Autosomno dominantni obrazac nasljeđivanja
Opisane kombinacije Noonan sindroma sa neurofibromatozom tipa I u nekoliko porodica sugerirale su moguću vezu između dva nezavisna lokusa 17q11.2 hromozoma 17. Neki pacijenti imaju mikrodelecije u 22q11 lokusu hromozoma 22; u ovim slučajevima kliničke manifestacije Noonan sindromi su u kombinaciji sa hipotireozom timusa i DiGeorgeovim sindromom. Brojni autori raspravljaju o uključenosti navodnih gena limfogeneze u patogenezu sindroma zbog prisustva facijalnih i somatskih anomalija sličnih Turnerovom sindromu i visoke incidencije patologije. limfni sistem.
Većina zajednički uzrok Noonan sindrom je mutacija gena PTPN11, koja se nalazi u otprilike 50% pacijenata. Protein kodiran genom PTPN11 pripada porodici molekula koji reguliraju odgovor eukariotskih stanica na vanjske signale. Najveći broj mutacije u Noonan sindromu su lokalizirane u egzonima 3,7 i 13 gena PTPN11, koji kodiraju proteinske domene odgovorne za prijelaz proteina u aktivno stanje.
Moguće ideje o patogenezi predstavljene su sljedećim mehanizmima:
RAS-MAPK put je veoma važan put transdukcije signala kroz koji prolaze ekstracelularni ligandi određene faktore rast, citokini i hormoni - stimulišu ćelijsku proliferaciju, diferencijaciju, preživljavanje i metabolizam (slika 2). Nakon vezivanja liganda, receptori na površini ćelije se fosforiliraju na mjestima njihove endoplazmatske regije. Ovo vezivanje uključuje adapterske proteine (npr. GRB2) koji formiraju konstitutivni kompleks sa faktorima razmjene gvanin nukleotida (npr. SOS) koji pretvaraju neaktivni GDP-vezani RAS u njegov aktivni GTP-vezani oblik. Aktivirani RAS proteini zatim aktiviraju RAF-MEKERK kaskadu kroz niz reakcija fosforilacije. Kao rezultat toga, aktivirani ERK ulazi u jezgro kako bi promijenio transkripciju ciljnih gena i korigirao aktivnost endoplazmatskih ciljeva kako bi inducirao adekvatne kratkoročne i dugoročne ćelijske odgovore na stimulus. Svi geni uključeni u Noonan sindrom kodiraju proteine sastavne za ovaj put i mutacije izazivaju bolesti, obično pojačavaju signal koji prolazi ovim putem.
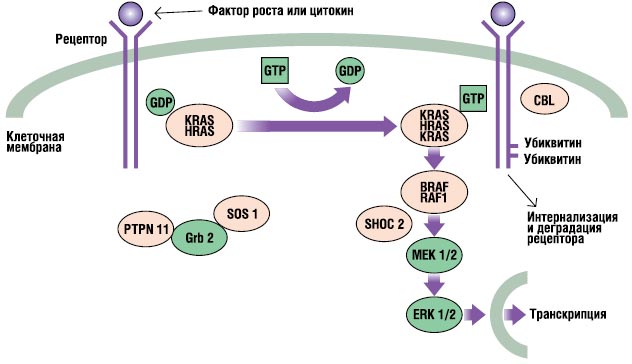
. RAS-MAPK signalni put. Signali rasta se prenose aktiviranim receptorima faktora rasta u jezgro. Mutacije u PTPN11, KRAS, SOS1, NRAS i RAF1 su povezane sa Noonan sindromom, a mutacije u SHOC2 i CBL su povezane sa fenotipom sličnim Noonan sindromu.
Kliničke karakteristike Noonan sindroma
Fenotip pacijenata sa Noonanovim sindromom podsjeća na Turnerov sindrom: kratak vrat s pterigoidnim naborom ili niskim rastom kose, niskim rastom, hipertelorizmom palpebralnih pukotina (slika 2). Mikroanomalije lica uključuju antimongoloidnu inciziju palpebralnih fisura, bočni kantus prema dolje, ptoza, epikantus, nisko ležeće ušne školjke, preklopljeni uvojak ušne školjke, malokluzija, rascjep uvule mehko nepce, gothic sky, micrognathia i microgenia. Grudni koš štitne žlijezde formira se sa hipoplastičnim široko razmaknutim bradavicama, sternum u gornjem dijelu strši, a u donjem tone. Oko 20% pacijenata ima umjereno tešku patologiju skeleta. Najčešći deformitet lijevka prsa, kifoza, skolioza; rjeđe - smanjenje broja vratnih kralježaka i njihova fuzija, nalik anomalijama u Klippel-Feil sindromu.
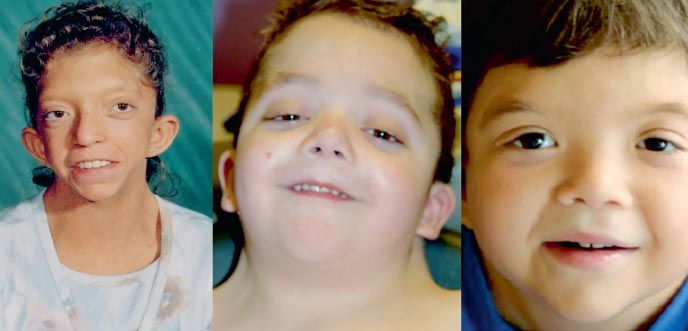
. Fenotip Noonanovog sindroma
Pacijenti sa Noonan sindromom obično imaju plavu, gustu, kovrdžavu kosu sa neobičnim rastom krune, često tamne mrlje na koži, hipertrihoza, degeneracija ploča nokta, anomalije u nicanju i rasporedu zuba, sklonost stvaranju keloidnih ožiljaka, povećana rastegljivost kože. Trećina pacijenata ima periferni limfedem, češće se limfedem šaka i stopala manifestuje kod dece rane godine. Čest znak je patologija vida (miopija, strabizam, umjereni egzoftalmus itd.). Zastoj u rastu javlja se kod približno 75% pacijenata, izraženiji je kod dječaka i obično je beznačajan. Zaostajanje u rastu manifestira se u prvim godinama života, rjeđe dolazi do blagog deficita u rastu i težini pri rođenju. Od prvih mjeseci života dolazi do smanjenja apetita. Starost kostiju obično zaostaje za dobi pasoša.
Karakteristična karakteristika sindroma je unilateralni ili bilateralni kriptorhizam, koji se javlja kod 70-75% muških pacijenata; kod odraslih pacijenata azoospermija, oligospermija, degenerativne promjene testisima. Ipak, pubertet se javlja spontano, ponekad sa određenim zakašnjenjem. Kod djevojčica često postoji kašnjenje u formiranju menstruacije, ponekad - kršenja menstrualnog ciklusa. Plodnost može biti normalna kod oba pola.
Mentalna retardacija se otkriva kod više od polovine pacijenata, obično manja. Često se primjećuju karakteristike ponašanja, dezinhibicija, poremećaj pažnje. Govor je obično bolje razvijen od ostalih intelektualne sfere. Stepen smanjenja inteligencije nije u korelaciji sa ozbiljnošću somatskih poremećaja [Marinčeva G.S., 1988]. U izolovanim slučajevima, malformacije centralnog nervni sistem(hidrocefalus, kile kičme), tromboembolijski infarkti mozga, moguće povezani sa vaskularnom hipoplazijom.
poroci unutrašnje organe sa Noonan sindromom su prilično karakteristični. Najtipičnije su kardiovaskularne anomalije: valvularna stenoza plućne arterije (oko 60% pacijenata), hipertrofična kardiomiopatija(20–30%), strukturne anomalije mitralni zalistak, atrijalni septalni defekti, tetralogija Fallot; koarktacija aorte je opisana samo kod muških pacijenata.
Kod trećine pacijenata evidentiraju se malformacije mokraćnog sistema (hipoplazija bubrega, udvostručenje karlice, hidronefroza, megaureter itd.).
Često se kod Noonanovog sindroma primjećuje pojačano krvarenje, posebno kod hirurške intervencije V usnoj šupljini i nazofarinksa. Nalaze se različiti defekti koagulacije: insuficijencija trombocitnog sistema, smanjenje nivoa faktora koagulacije, posebno XI i XII, povećanje tromboplastinskog vremena. Postoje izvještaji o kombinaciji Noonanovog sindroma s leukemijom i rabdomiosarkomom, što može ukazivati na blagi porast rizika od maligniteta kod ovih pacijenata.
U tabeli 1 prikazane su karakteristike fenotipa u Noonan sindromu, koje se mijenjaju s godinama bolesnika. Tabela 2 pokazuje korelaciju između fenotipa i genotipa u Noonan sindromu.
Tabela 1. Tipične crte lica kod pacijenata sa Noonan sindromom prema godinama
| Čelo, lice, kosa | Oči | Uši | Nos | Usta | Vrat | |
| novorođenče* | Visoko čelo, niska linija kose u potiljačnoj regiji | Hipertelorizam, nadole palpebralne pukotine, epikantalni nabor | – | Kratak i širok udubljen korijen, okrenut vrh | Duboko udubljen filtrum, visoki široki vrhovi crne ivice usana, mikrognatija | Višak kože na potiljku |
| Grudi (2-12 mjeseci) | Velika glava, visoko i izbočeno čelo | Hipertelorizam, ptoza ili debeli spušteni kapci | – | Kratak i širok udubljeni korijen | – | – |
| Dijete (1-12 godina) | Grube crte lica, dugo lice | – | – | – | – | – |
| Tinejdžer (12-18 godina) | Miopatsko lice | – | – | Most je visok i tanak | – | Očigledno formiranje nabora na vratu |
| Odrasli (>18 godina) | Prepoznatljive crte lica su profinjene, koža djeluje tanko i prozirno | – | – | Izbočeni nasolabijalni nabor | – | – |
| Svi uzrasti | – | Plave i zelene šarenice, obrve u obliku dijamanta | Niske, unazad zakrenute uši sa debelim naborima | – | – | – |
tabela 2. Korelacije između genotipa i fenotipa u Noonan sindromu*
| Kardiovaskularni sistem | Visina | Razvoj | Koža i kosa | Ostalo | |
| PTPN11 (cca. 50%) | Stenoza plućnog trupa je izraženija; manje - hipertrofična kardiomiopatija i defekt atrijalnog septuma | Niži rast; niža koncentracija IGF1 | Pacijenti sa N308D i N308S imaju blagi pad ili normalnu inteligenciju | – | Izraženija hemoragijska dijateza i juvenilna mijelomonocitna leukemija |
| SOS1 (približno 10%) | Manji defekt atrijalnog septuma | Veći rast | Manji pad inteligencije, odložen razvoj govora | Slično kardiokutanom sindromu lica | – |
| RAF1 (cca. 10%) | Teža hipertrofična kardiomiopatija | – | – | Više madeži, lentigo, fleke od kafe sa mlekom | – |
| KRAS (<2%) | – | – | Ozbiljnije kognitivno kašnjenje | Slično kardio-kožnom sindromu lica | – |
| NRAS (<1%) | – | – | – | – | – |
Podaci iz laboratorijskih i funkcionalnih studija
Ne postoje specifični biohemijski markeri za dijagnozu Noonan sindroma. Kod nekih pacijenata, smanjenje spontanog noćnog lučenja hormona rasta uz normalan odgovor na farmakološke stimulacijske testove (klofelin i arginin), smanjenje nivoa somatomedina-C i smanjenje odgovora somatomedina na uvođenje hormona rasta su otkriveni.
Dijagnostički kriteriji
Dijagnoza "Noonan sindrom" postavlja se na osnovu kliničkih znakova, u nekim slučajevima dijagnoza se potvrđuje rezultatima molekularno-genetske studije. Kriterijumi za dijagnosticiranje sindroma uključuju prisustvo karakterističnog lica (sa normalnim kariotipom) u kombinaciji sa jednom od sljedećih karakteristika: srčana bolest, nizak rast ili kriptorhizam (kod dječaka), odgođeni pubertet (kod djevojčica). Za otkrivanje kardiovaskularne patologije potrebno je provesti ultrazvučni pregled srca s dinamičkim određivanjem veličine šupljina i zidova ventrikula. Prenatalna dijagnoza bolesti moguća je uz pomoć ultrazvučnog praćenja, što omogućava otkrivanje srčanih mana i anomalija u strukturi vrata.
Diferencijalna dijagnoza
Kod djevojčica se diferencijalna dijagnoza postavlja prvenstveno s Turnerovim sindromom; Dijagnoza se može razjasniti citogenetskim pregledom. Fenotipski znaci Noonan sindroma nalaze se i kod niza drugih bolesti: Williamsov sindrom, LEOPARD sindrom, Dubovitz, kardiofacio-kutani sindrom, Cornelia de Lange, Cohen, Rubinstein-Taybi, itd. molekularne genetičke studije svakog sindroma sa značajnim kliničkim materijalom koji se trenutno aktivno razvija.
Tretman
Liječenje pacijenata sa Noonan sindromom usmjereno je na otklanjanje defekata kardiovaskularnog sistema, normalizaciju mentalnih funkcija, stimulaciju rasta i seksualnog razvoja. Za liječenje pacijenata s displazijom zalistaka plućne arterije, između ostalih metoda, uspješno se koristi i balon valvuloplastika. Za stimulaciju mentalnog razvoja koriste se nootropni i vaskularni agensi. Lijekovi usmjereni na stimulaciju seksualnog razvoja indicirani su uglavnom za pacijente s kriptorhizmom. Preparati horionskog gonadotropina se koriste u starosnim dozama. U starijoj dobi - u prisustvu hipogonadizma - preparati testosterona. Poslednjih godina, rekombinantni oblici humanog hormona rasta koriste se u lečenju pacijenata sa Noonan sindromom. Klinički podaci su potvrđeni povećanjem nivoa somatomedina-C i specifičnog vezujućeg proteina tokom terapije. Konačna visina pacijenata koji primaju dugotrajnu terapiju hormonom rasta u nekim slučajevima premašuje prosječnu visinu članova porodice.
Prognoza za život je određen ozbiljnošću kardiovaskularne patologije.
Prevencija bolest se zasniva na podacima medicinskog genetičkog savjetovališta.
Medicinsko genetičko savjetovanje
U medicinsko-genetičkom savjetovanju treba polaziti od autosomno dominantnog tipa nasljeđivanja i visokog (50%) rizika od recidiva bolesti u porodici sa nasljednim oblicima. Da bi se utvrdila priroda vrste nasljeđivanja, potrebno je provesti temeljit pregled roditelja, jer se sindrom može manifestirati s minimalnim kliničkim simptomima. Trenutno je razvijena i unapređuje se molekularna genetička dijagnoza bolesti tipizacijom mutacija u genima: PTPN11, SOS1, RAF1, KRAS, NRAS itd. Razvijaju se metode prenatalne dijagnostike bolesti.
Kliničko posmatranje
Dječak G., 9 godina (slika 3), promatran je u mjestu stanovanja od strane genetičara sa dijagnozom hromozomske patologije?, Williamsov sindrom (poseban fenotip, zadebljanje kvržica mitralnog zaliska, hiperkalcemija jednom u 3 godine) ?.
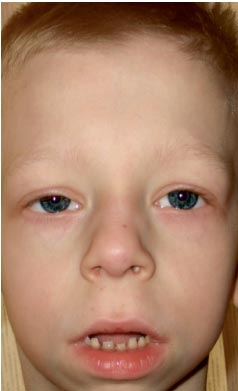
. Osobitosti fenotipa djeteta s Noonan sindromom (izduženi kostur lica s „bucmastim obrazima“, kratak vrat, kriloidni nabori na vratu, skraćeni nos sa nozdrvama otvorenim naprijed, natečene usne, nagnuta brada, antimongoloid incizija palpebralnih fisura, malokluzija, makrostomija)
Pritužbe na smanjenu memoriju, umor, smanjenu brzinu rasta.
Porodična historija : roditelji su Rusi po nacionalnosti, nisu u krvnom srodstvu i nemaju profesionalnih opasnosti, zdravi. Visina oca je 192 cm, visina majke je 172 cm.U pedigreu slučajeva mentalnih bolesti, epilepsije, zastoja u razvoju nisu zabeleženi.
Istorija života i bolesti : dječak iz 2. trudnoće (1. trudnoća - m/a), koji je cijelo vrijeme tekao uz prijetnju prekida, praćen polihidramniom. Porođaj je bio prvi, na vrijeme, brz, porođajna težina - 3400 g, dužina - 50 cm. Odmah je vrisnuo, Apgar ocjena - 7/9 bodova. Prilikom rođenja, neonatolog je skrenuo pažnju na neobičan fenotip djeteta, preporučio proučavanje kariotipa, rezultat je 46, XY (normalan muški kariotip). Sumnja se na kongenitalnu hipotireozu, urađena je studija profila štitnjače, rezultat je normalan status štitnjače. Nadalje, dijete je promatrao genetičar sa pretpostavljenom dijagnozom "Vilijamsov sindrom". Rani postnatalni period - bez osobina. Motorički razvoj po godinama, prve riječi - po godini, frazni govor - sa 2 godine 3 mjeseca.
U dobi od 8 godina, konsultovao ga je endokrinolog zbog smanjenog rasta, umora i smanjene memorije. Rendgenskim pregledom šaka utvrđeno je umjereno zaostajanje u koštanom dobu (BC) u odnosu na pasoško (BC odgovara 6 godina). Proučavanje profila štitnjače otkrilo je umjereno povećanje tireostimulirajućeg hormona uz normalan nivo slobodnog T4 i drugih pokazatelja; Ultrazvuk štitne žlijezde - bez patologije. Propisana je hormonska terapija, praćena dinamičkom opservacijom.
Uzimajući u obzir nesigurnost dijagnoze u mjestu stanovanja, genetičar je dijete uputio u Moskovski regionalni konsultativno-dijagnostički centar za djecu kako bi se razjasnila dijagnoza.
Podaci objektivnog istraživanja:
Visina - 126 cm, težina - 21 kg.
Fizički razvoj ispodprosječan, skladan. Sds rasta odgovara -1 (normalno -2 + 2). Karakteristike fenotipa (fotografija 3): izduženi kostur lica sa „bucmastim obrazima“, kratak vrat, kriloidni nabori na vratu, niska dlakavost na vratu, kratak nos sa otvorenim nozdrvama, natečene usne, nagnuta brada, antimongoloidni rez palpebralne pukotine, malokluzija, makrostomija, hipertelorizam bradavica, asimetrija grudnog koša, nepotpuna sindaktilija kože 2. ili 3. prsta na stopalima, izražena hipermobilnost interfalangealnih zglobova, lomljivi, suhi nokti. Na unutrašnjim organima - bez karakteristika. Seksualni razvoj - Tanner I (koji odgovara prepubertetskom periodu).
Podaci iz laboratorijskih i funkcionalnih studija:
Klinička analiza krvi i urina je norma.
Biohemijska analiza krvi - indikatori u granicama normale.
Profil štitnjače (TSH) - 7,5 μIU / ml (normalno - 0,4-4,0), ostali pokazatelji su normalni.
Somatotropni hormon (STH) - 7 ng / ml (norma - 7-10), somatomedin-C - 250 ng / ml (norma - 88-360).
Ultrazvuk štitne žlijezde - bez patologije.
Ultrazvuk unutrašnjih organa - bez karakteristika.
EKG - sinusna tahikardija, normalan položaj električne ose srca.
Ehokardiografija - MVP 1. stepena sa minimalnom regurgitacijom, miksomatoznim zadebljanjem kvržica mitralnog zaliska, dodatnim akordom u šupljini lijeve komore.
R-grafija kičme - desnostrana skolioza torakalne kičme I stepen.
R-grafija šaka sa zahvatom podlaktica - koštano doba 7-8 godina.
EEG obrasci epileptičke aktivnosti nisu registrovani.
MRI mozga - bez patoloških promjena.
Audiogram - bez patologije.
DNK dijagnostika: molekularna genetska studija - nisu otkrivene delecije proučavanih lokusa kritične regije hromozoma 7; Gly434Ary (1230G>A) mutacija je pronađena u 11. egzonu gena SOS1 (analiza gena PTPN11 - nisu pronađene mutacije), što je tipično za Noonan sindrom.
Savjet strucnjaka:
Endokrinolog- subklinički hipotireoza, nepotpuna kompenzacija lijeka.
Optometrista- astigmatizam.
Neurolog- vegetativna distonija. neurotične reakcije.
Kardiolog- funkcionalna kardiopatija.
Ortopedski hirurg- kršenje držanja. Deformitet grudnog koša.
Genetičar Noonan sindrom.
Uzimajući u obzir fenotip djeteta, podatke iz anamneze, rezultate dodatnih studija, postavljena je dijagnoza Noonan sindroma, što je potvrđeno rezultatom molekularne genetske studije.
Dakle, prikazano kliničko zapažanje pokazuje složenost diferencijalno-dijagnostičke pretrage, potrebu integrisanja pojedinačnih znakova u opći fenotip određenog patološkog stanja radi ciljane pravovremene dijagnoze pojedinih oblika nasljednih bolesti, te važnost molekularno-genetskih metoda za razjašnjavanje dijagnoza. Pravovremena dijagnoza, razjašnjenje geneze svakog sindroma posebno su važni, jer vam omogućavaju da pronađete najbolji pristup liječenju ovih stanja, prevenciji mogućih komplikacija (sve do invaliditeta djeteta); prevencija recidiva nasljednih bolesti u oboljelim porodicama (medicinsko genetičko savjetovanje). To diktira potrebu da liječnici različitih specijalnosti jasno upravljaju tokom nasljedne patologije.
Bibliografija:
- Baird P., De Jong B. Noonanov sindrom (XX i XY Turnerov fenotip) u tri generacije porodice // J. Pediatr., 1972, vol. 80, str. 110–114.
- Hasegawa T., Ogata T. et al. Koarktacija aorte i bubrežna hupoplazija kod dječaka s Turner/Noonan površinskim anomalijama i kariotipom 46, XY: klinički model za moguće oštećenje pretpostavljenog limfogenog gena(ova) za Turnerove somatske stigme // Hum. Genet., 1996, vol. 97, str. 564–567.
- Fedotova T.V., Kadnikova V.A. et al. Kliničko-molekularno-genetička analiza Noonanovog sindroma. Materijali VI kongresa Ruskog društva medicinske genetike. Medicinska genetika, Dodatak br. 5, 2010, str.184.
- Ward K.A., Moss C., McKeown C. Kardio-facio-kutani sindrom: manifestacija Noonan sindroma? // Br. J. Dermatol., 1994, vol. 131, str. 270–274.
- Municchi G., Pasquino A.M. et al. Liječenje hormonom rasta u Noonan sindromu: izvještaj o četiri slučaja koji su dostigli konačnu visinu // Horm. Res., 1995, vol. 44, str. 164–167.
Kod djece postoje različite urođene patologije. Neki od njih se smatraju prilično čestim. Ali postoje i rijetke bolesti. Njihova prevalencija je niska. Međutim, mnogi imaju prilično ozbiljne posljedice. Jedna od ovih patologija je Noonanov sindrom. Zatim, razmotrite to detaljnije. Članak će opisati kako i zašto se pojavljuje Noonanov sindrom. U tekstu će biti predstavljena i fotografija bolesti.
Opće informacije
Noonanov sindrom je nasljedna patologija. Mutirani gen PTPN11 od roditelja koji ga nose prenosi se na potomstvo. Muškarci su po pravilu neplodni. Stoga se gen prenosi po majčinoj liniji. Obično se navodi porodična priroda ove bolesti. Rijetki slučajevi koji nisu vezani za nasljeđe, međutim, zabilježeni su iu praksi.
Istorijska referenca
Svojevremeno je Jacqueline Noonan, koja se bavila pedijatrijskim kardiologom, dok je radila na klinici na Univerzitetu u Ajovi, primijetila da se određena grupa djece, među kojima su i dječaci i djevojčice sa plućnom stenozom, često razlikovala po niskom rastu, razrogačenih očiju, prepletenih vratova, niskih ušnih školjki i ptoze. Nakon ispitivanja kombinacije manifestacija srčanih oboljenja sa drugim razvojnim anomalijama kod 833 pacijenta, doktor je 1962. godine napisao članak. U svom radu opisala je devet slučajeva u kojima su uočene karakteristične crte lica i nizak rast na pozadini kongenitalnog tipa. Bolest je otkrivena i kod muškaraca i kod žena. 
Noonanov sindrom: simptomi
Postoji niz karakterističnih karakteristika koje razlikuju patologiju od drugih. To uključuje, posebno:
- Niska visina. Za žene - 1,53, za muškarce - 1,63 m. U trenutku rođenja i dužina tijela i težina djeteta su u granicama normale. Zastoj u rastu počinje u dobi od 2-3 godine.
- Promjene na licu. Pacijenti s dijagnozom Noonanovog sindroma imaju široko razmaknute oblike. U unutrašnjem uglu se nalazi kožni nabor. Postoji i ptoza (spušteni kapci) zbog oslabljene mišićne funkcije ili zbog malih očnih duplji. Otprilike dvije trećine sve djece sa patologijom ima strabizam.Kada je ptoza ograničena i razvija se strabizam.
- Povrede strukture čeljusti. Gornji je nerazvijen, ima lučno visoko nepce. U obje čeljusti položaj zuba je nepravilan.
- Nisko slijetanje ili deformacija ušnih školjki. Kao rezultat ove anomalije, sluh je oštećen.
- Kratak i širok vrat.
- Štit grudi sa široko razmaknutim bradavicama.
- Deformitet u zglobu lakta (kongenitalni).
- Ravna stopala.
- Kratki prsti.
Malformacije unutrašnjih organa
Noonan sindrom najčešće prate poremećaji u radu kardiovaskularnog sistema. U pozadini patologije, otkriva se suženje (stenoza) u plućnom deblu, kao i defekt interventrikularnog septuma. Drugo mjesto zauzimaju anomalije genitourinarnog sistema. Dakle, na dijelu bubrega otkrivaju se defekti kao što su hipoplazija (nedostatak razvoja tkiva) ili nedostatak jednog bubrega. Što se tiče puberteta, on varira od normalnog do potpuno defektnog. Djevojčice često imaju kasnu menstruaciju, dječaci imaju kriptorhizam ili su testisi potpuno odsutni. Postoje i poremećaji spermatogeneze. U nekim slučajevima spermatozoidi su potpuno odsutni. U pozadini operacija s Noonanovim sindromom, zabilježeno je pojačano krvarenje. Neki pacijenti mogu pokazati i blage abnormalnosti. 
Forms
Postoje dvije vrste patologije:
- porodični oblik. Razlikuje se po nasljednoj prirodi autosomno dominantnog tipa. Kod nosilaca mutantnog gena, potomci se pojavljuju s patologijom.
- sporadični oblik. U ovom slučaju, mutacija se pojavljuje od slučaja do slučaja. U ovom slučaju nije utvrđen nasljedni faktor.
Uzroci
Najčešći faktor koji izaziva patologiju je mutacija gena PTPN11. Ovaj uzrok se otkriva kod 50% pacijenata. Međutim, kod određenog procenta osoba sa ovim sindromom genetski faktor je nejasan. Nasljeđivanje patologije javlja se u autosomno dominantnom obliku. Sindrom može biti izazvan novom mutacijom. Iz ovoga proizilazi da roditelji sa takvim genom nemaju velike šanse da dobiju još jedno dijete.
Dijagnoza
Instalira se u skladu sa karakterističnim vanjskim karakteristikama (gore su opisane). Laboratorijski indikatori se takođe ispituju tokom dijagnoze. Konkretno, dolazi do smanjenja koncentracije testosterona, faktora koagulacije XII. Izvode se i instrumentalne studije. Propisan je rendgenski snimak grudne kosti, ehokardiografija, EKG. Uz pomoć ovih metoda otkrivaju se anomalije unutrašnjih organa. Lekar takođe može zakazati konsultacije 
Terapeutske aktivnosti
Zbog genetskog stanja sindroma, liječenje je uglavnom usmjereno na uklanjanje simptoma. Kod kriptorhizma je propisana operacija. Tokom nje, testisi se kreću u skrotum. U pozadini smanjenja koncentracije androgena preporučuje se hormonska terapija. U prisustvu zatajenja bubrega propisuje se hemodijaliza. Uz njegovu pomoć, proizvodi metaboličkih procesa uklanjaju se iz tijela kroz ekstrarenalno pročišćavanje krvi.
Noonan sindrom(J.A. Noonan, američki endokrinolog, rođen 1928.) je nasledna bolest koju karakterišu niski rast i anomalije u somatskom razvoju. J. Nunen je najpotpunije opisan 1963. Može biti sporadičan ili porodičan, naslijeđen na autosomno recesivan način. Patološki promijenjeni geni koji uzrokuju N. stranice lokalizirani su u autosomima (vidi. nasljedne bolesti ). Spolni hromatin i kariotip hromozomi ) odgovaraju spolu pacijenta. Bolest se javlja i kod muškaraca i kod žena.
Glavne kliničke manifestacije N. sa. su nizak rast, donji ekstremiteti, pterigoidni nabori kože na vratu, niska linija kose na potiljku, karakteristične dermatoglifske promjene, hipertelorizam, ptoza, antimongoloidna incizija oka, deformitet ili slabo prianjanje naušnih rubova, asimetrija lice, široki nos, što pacijente čini sličnima jedni drugima. prijatelj ( pirinač .). Karakterizira ga kratak vrat, urođeni deformitet zgloba lakta (cubitus valgus), brahidaktilija, visoko nepce, patološki ugristi , ravna stopala, promjene oblika i strukture pršljenova i druge anomalije uočene tokom Shereshevsky-Turnerov sindrom . Često se nalazi stenoza plućnog trupa, defekt ventrikularnog septuma. Druge kongenitalne malformacije kardiovaskularnog sistema (vidi. Urođene srčane mane ) su manje uobičajene. Seksualni razvoj na N. str. varira od normalne do potpune disgeneze gonada (vidi Hermafroditizam ). Djevojčice često imaju kasnu menstruaciju, dječaci - kriptorhizam , hipogonadizam . Histološkim pregledom materijala dobivenog biopsijom testisa otkriva se smanjenje ili potpuno odsustvo zametnih stanica i hiperplazija Leydigovih stanica. Ostale manifestacije N. sa. su ginekomastija i mentalna retardacija. U krvnom serumu pacijenata, sadržaj testosterona, estrogena (vidi. polni hormoni ) i gonadotropina (vidi hormoni hipofize ) je normalna ili promijenjena u zavisnosti od težine gonadne disgeneze. Koncentracija hormona rasta u krvi je normalna. Kariotip 46XX ili 46XV odgovara spolu gonade i pasoša.
Dijagnoza se postavlja na osnovu karakterističnih kliničkih znakova i laboratorijskih podataka. N. s. najčešće se diferencira sa Shereshevsky-Turnerov sindromom ( sto .).
Liječenje u prisustvu znakova hipogonadizma sastoji se u zamjenskoj terapiji preparatima polnih hormona. Upotreba hormona rasta je neefikasna. Prema indikacijama, hirurška korekcija kongenitalnih malformacija, liječenje mentalnih poremećaja povezanih s mentalnom retardacijom (vidi. Endokrini mentalni poremećaji ). Prognoza za život je povoljna. Posebnu pažnju treba obratiti na zapošljavanje: izbor specijalnosti treba da doprinese socijalnoj adaptaciji pacijenata, uzimajući u obzir njihove mentalne sposobnosti i fizičke nedostatke.
Diferencijalni dijagnostički znakovi Noonanovog i Shereshevsky-Turnerovog sindroma
klinički znak |
Noonan sindrom |
Shereshevsky-Turnerov sindrom |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
seksualni razvoj |
Noonan sindrom karakteriziraju višestruke kongenitalne malformacije tipične za pacijente sa Shereshevsky-Turner sindromom, s normalnim kariotipom. Ponekad se sindrom identificira sa Bonnevie-Ulrichovim sindromom, koji ima sličnu kliničku sliku. Međutim, kod Bonnevie-Ulrichovog sindroma nema srčane bolesti, koja se vrlo često uočava kod Shereshevsky-Turnerovog sindroma i Noonan. Kod svih pacijenata mogu se otkriti manje ili više izraženi simptomi hipogonadizma, iako neki autori smatraju da se kod Noonanovog sindroma genitalije kod nekih dječaka normalno razvijaju. Etiologija. Sindrom se nasljeđuje na autosomno dominantan način sa niskom penetracijom gena. Distribucija sindroma je prilično velika (1:1000 - 1:5000), ali ne postoji jasan dijagnostički kriterij u obliku abnormalnog kariotipa, kao kod pacijenata sa Shereshevsky-Turnerovim sindromom. S tim u vezi, ne otkrivaju se svi slučajevi bolesti. Odnosi hipotalamus-hipofiza-gonada (patogeneza) u Noonan sindromu praktički nisu proučavani. Postoji samo jedan izvještaj o karakteristikama reproduktivnog sistema kod pacijenata, na osnovu kojih je teško napraviti generalizacije. Ispitivali smo sadržaj testosterona i gonadotropnih hormona u krvi, kao i funkcionalne rezerve testisa kod 8 dječaka s Noonan sindromom. Nivo testosterona kod svih pacijenata bio je ispod normalnog. Izlučivanje 17-KS urinom je također smanjeno. Ovi nalazi ukazuju na hormonalni nedostatak testisa u Noonan sindromu. Nivo testosterona i gonadotropnih hormona u serumu i izlučivanje mokraćom od 17KS (M±m) kod pacijenata sa Noonan sindromom
Kombinacija nedovoljnog lučenja testosterona i visokog nivoa LH kod 4 pacijenta ukazuje na primarnu leziju testisa, uglavnom intersticijskih ćelija. Međutim, primjena CG tokom 3 dana u dnevnoj dozi od 1500 IU/m2 dovela je do značajnog povećanja nivoa testosterona u krvi kod ovih dječaka. Kauschunsky et al. (1977) kada su davali GnRH pacijentu sa Noonan sindromom, uočili su izraženo povećanje lučenja gonadotropina, kao kod pacijenata sa primarnim hipogonadizmom, i istovremeno pozitivnu reakciju testisa na CG. Ograničen broj zapažanja otežava tumačenje podataka. Može se pretpostaviti da je kod pacijenata smanjena osjetljivost receptora Leydigovih stanica na endogeni gonadotropin, ali su rezerve testisa očuvane, što se izražava u povećanju lučenja testosterona nakon stimulacije visokim dozama hCG. Kod pacijenata sa niskim nivoom testosterona i normalnim nivoom gonadotropnih hormona u krvi, patogeneza insuficijencije testisa verovatno je bliska patogenezi normogonadotropnog hipogonadizma. Theintz, Savagl (1982) opisao je 5 dječaka od 11,8 do 16,5 godina sa Noonan sindromom. Svi su imali jasne znakove hipogonadizma i zaostajali u seksualnom razvoju. Kod njih 4, tada se polni razvoj normalizirao, a kod dvoje spontano, a kod dvoje - nakon tretmana testosteronom. Samo kod jednog pacijenta liječenje CG i testosteronom bilo je neefikasno. Dakle, u bolesnika s Noonan sindromom, unatoč zajedničkoj kliničkoj slici, priroda hipogonadizma može biti drugačija. Insuficijencija reproduktivnog sistema kod njih može biti posljedica primarnog defekta gonada, disfunkcije hipotalamo-hipofiznog sistema i promjene osjetljivosti testisa na gonadotropnu stimulaciju. Klinička slika podsjeća na kliniku Shereshevsky-Turnerovog sindroma. Evo izvoda iz istorije bolesti jednog od pacijenata koje smo posmatrali. Sereža L., stara 8 godina, primljena je na pedijatrijsku endokrinologiju bolnice. Rauhfus 05.03.82. sa zaostatkom u fizičkom i mentalnom razvoju. Rođen je iz druge trudnoće, koja je prošla sa teškom toksikozom. U 7. - 8. nedelji trudnoće majka je pretrpela akutnu respiratornu virusnu infekciju. Prva trudnoća završila se pobačajem. Majka pati od urođene Dječak je rođen prijevremeno (vađen carskim rezom). Telesna težina 3500 g, dužina 51 cm U neonatalnom periodu stanje je teško. U dobi od 4 mjeseca dijagnosticirana je srčana bolest; dječak je zaostajao u fizičkom i mentalnom razvoju od prve godine. Odsustvo testisa u skrotumu otkriveno je pri rođenju. Prilikom prijema dužina tijela 124 cm (ispod prosjeka), težina 22 kg. Fizik je nepravilan - širok kratak vrat, asimetrična prsa, deformitet grudne kosti u obliku lijevka; visoko nepce, hipertelorizam, deformitet uha, defekti prstiju, široko razmaknute bradavice. Penis je zadovoljno razvijen, skrotum je veoma mali, testisi nisu opipljivi. Inteligencija je umjereno smanjena. Karyotype 46XY. Rendgen lubanje bez obilježja. Starost kostiju odgovara 7 godina. U krvnom serumu LH 4,33 mIU / ml, FSH 3,13 mIU / ml, testosteron 0,56 nmol / l (svi pokazatelji su smanjeni). Izlučivanje 17KS 9,1 mmol/dan. Funkcionalni test sa jednom injekcijom hCG je negativan - testosteron u krvi - 0,57 nmol/l, 17-KS - 8,87 mmol/dan; 3-dnevni test je pozitivan - testosteron - 1,54 nmol / l, 17KS - 10,15 mmol / dan. Na osnovu anamneze, kliničke slike i laboratorijskih podataka postavljena je dijagnoza Noonanovog sindroma. Pozitivan 3-dnevni test je omogućio liječenje CG. U nedostatku efekta indicirano je kirurško liječenje kriptorhizma. "Poremećaji seksualnog razvoja kod dječaka", Izgled.
Forms
Uzroci
DijagnostikaDijagnoza se postavlja na osnovu:
Liječenje Noonan sindroma
Komplikacije i posljedice
Prevencija Noonan sindroma
Slični članci
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||