Noonan-szindróma és hasonló betegségek. Nézze meg, mi az a „Noonan-szindróma” más szótárakban.
E.V. Tozliyan, gyermek endokrinológus, genetikus, Ph.D., Külön szerkezeti felosztás"Klinikai Kutatóintézet Gyermekgyógyászat" Állami Költségvetési Felsőoktatási Szakmai Oktatási Intézmény Orosz Nemzeti Kutató Orvostudományi Egyetem névadója. N.I. Pirogov, az Orosz Föderáció Egészségügyi Minisztériuma, Moszkva Kulcsszavak: gyerekek, Noonan szindróma, diagnózis.
Kulcsszavak: gyerekek, Noonan szindróma, diagnosztika.
A cikk ismerteti a Noonan-szindrómát (Ullrich-Noonan szindróma, turneroid szindróma normál kariotípussal) - egy ritka veleszületett patológia, autoszomális domináns módon öröklődik, hordoz családi karakter Előfordulnak azonban szórványos esetek is. A szindróma feltételezi a Shereshevsky-Turner szindrómára jellemző fenotípus jelenlétét normál kariotípusú női és férfi egyénekben. Által benyújtott klinikai megfigyelés. A differenciáldiagnosztikai keresés nehézségei és a klinikusok tájékozottságának hiánya a ez a szindrómaés az interdiszciplináris megközelítés fontossága.
Történelmi tények
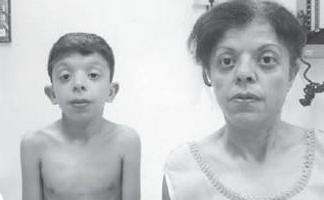
Először kb szokatlan szindróma O. Kobylinski említi 1883-ban (1. fotó).
A legrégebbi ismert klinikai eset Noonan-szindróma, amelyet 1883-ban ír le O. Kobylinski
A betegséget 1963-ban írta le Jacqueline Noonan amerikai kardiológus, aki kilenc billentyűszűkületben szenvedő betegről számolt be. pulmonalis artéria, alacsony termet, hypertelorizmus, mérsékelt intelligenciacsökkenés, ptosis, kriptorchidizmus és csontrendszeri rendellenességek. Dr. Noonan, aki mint gyermek kardiológus az Iowai Egyetemen észrevette, hogy a gyerekek a ritka típus szívelégtelenség - pulmonalis billentyűszűkület - gyakran figyeltek meg olyan tipikus testi rendellenességeket, mint az alacsony termet, a szárnyas nyak, a tágra fektetett szemek és az alacsonyan fektetett fülek. A fiúk és a lányok egyaránt érintettek. Dr. Opitz János, volt diák Noonan volt az első, aki megalkotta a „Noonan-szindróma” kifejezést, amely a Noonan által leírtakhoz hasonló tüneteket mutató gyermekek állapotát írja le. Noonan később megírta a „Hypertelorism with Turner Phenotype” című cikket, és a „Noonan-szindróma” nevet 1971-ben hivatalosan elismerték a szív- és érrendszeri betegségekről tartott szimpóziumon.
Etiológia és patogenezis
A Noonan-szindróma egy autoszomális domináns rendellenesség, változó expresszivitással (1. ábra). A Noonan-szindróma génje lokalizálódik hosszú váll 12-es kromoszóma. A szindróma genetikai heterogenitása nem zárható ki. Leírták a szindróma szórványos és családi formáit, autoszomális domináns öröklődési formával. Családi ügyekben mutáns gén rendszerint az anyától öröklődik, mivel súlyos fejlődési rendellenességek miatt urogenitális rendszer az ilyen állapotú férfiak gyakran terméketlenek. A legtöbb jelentett eset szórványos, de novo mutációk okozzák.
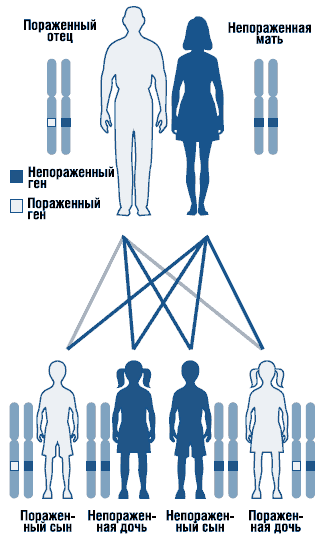
. Autoszomális domináns öröklődési típus
A Noonan-szindróma és az I. típusú neurofibromatózis leírt kombinációi több családban arra utaltak, hogy a 17. kromoszóma két független 17q11.2 lókusza között lehetséges kapcsolat. Egyes betegeknél a 22. kromoszóma 22q11 lókuszában mikrodeléciót észleltek; ezekben az esetekben klinikai megnyilvánulásai A Noonan-szindrómát a csecsemőmirigy alulműködésével és a DiGeorge-szindrómával kombinálják. Számos szerző tárgyalja a feltételezett limfogenezis gének részvételét a szindróma patogenezisében a Turner-szindrómához hasonló arc- és szomatikus anomáliák jelenléte és a patológia magas gyakorisága miatt. nyirokrendszer.
A legtöbb gyakori ok A Noonan-szindróma a PTPN11 gén mutációja, amely a betegek körülbelül 50%-ában található meg. A PTPN11 gén által kódolt fehérje olyan molekulák családjába tartozik, amelyek szabályozzák az eukarióta sejtek külső jelekre adott válaszát. Legnagyobb szám A Noonan-szindróma mutációi a PTPN11 gén 3., 7. és 13. exonjában lokalizálódnak, amelyek a fehérje aktív állapotba való átmenetéért felelős fehérjedoméneket kódolnak.
A patogenezissel kapcsolatos lehetséges elképzeléseket a következő mechanizmusok képviselik:
A RAS-MAPK útvonal egy nagyon fontos jelátviteli út, amelyen keresztül az extracelluláris ligandumok - bizonyos tényezők növekedés, citokinek és hormonok – serkentik a sejtproliferációt, a differenciálódást, a túlélést és az anyagcserét (2. ábra). A ligandum megkötésekor a sejtfelszíni receptorok foszforileződnek az endoplazmatikus régiójukban. Ez a kötődés adapterfehérjéket (pl. GRB2) foglal magában, amelyek konstitutív komplexet képeznek a guanin nukleotid cserélő faktorokkal (pl. SOS), amelyek az inaktív GDP-hez kötött RAS-t az aktív GTP-kötött formává alakítják. Az aktivált RAS fehérjék ezután egy sor foszforilációs reakción keresztül aktiválják a RAF-MEKERK kaszkádot. Ennek eredményeként az aktivált ERK belép a sejtmagba, hogy megváltoztassa a célgének transzkripcióját, és beállítja az endoplazmatikus célpontok aktivitását, hogy megfelelő rövid és hosszú távú sejtválaszokat indukáljon az ingerekre. A Noonan-szindrómában részt vevő összes gén ennek az útvonalnak a szerves részét képező fehérjéket és mutációkat kódol betegséget okozva, általában felerősítik az ezen az úton áthaladó jelet.
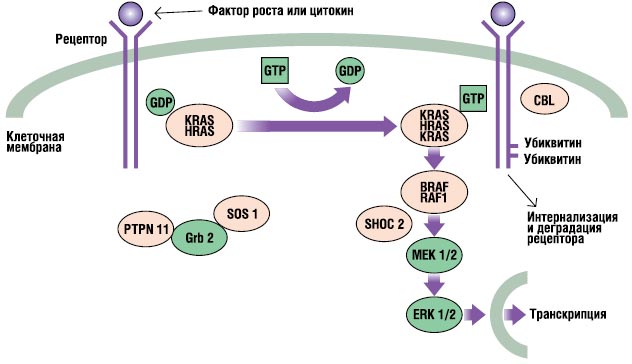
. RAS-MAPK jelátviteli útvonal. A növekedési jelek a növekedési faktor által aktivált receptoroktól a sejtmagba jutnak. A PTPN11, KRAS, SOS1, NRAS és RAF1 mutációi Noonan-szindrómával, a SHOC2 és CBL mutációi pedig Noonan-szindrómaszerű fenotípussal járnak
A Noonan-szindróma klinikai jellemzői
A Noonan-szindrómás betegek fenotípusa a Turner-szindrómához hasonlít: rövid nyak pterigoid redővel vagy alacsony szőrnövekedéssel, alacsony testalkattal, a palpebralis repedések hypertelorizmusával (2. fotó). Az arc mikroanomáliái közé tartoznak az antimongoloid palpebrális repedések, a lefelé tartó külső canthusok, a ptosis, az epicanthus, az alacsonyan elhelyezkedő fülkagylók, a hajtogatott hélix fülek, rossz elzáródás, uvula hasadék puha szájpadlás, gótikus szájpadlás, mikrognathia és mikrogénia. A mellkas pajzsmirigy alakú, hipoplasztikus, szélesen elhelyezkedő mellbimbókkal, a szegycsont felső részen kinyúlik, alul lesüllyed. A betegek körülbelül 20% -ánál mérsékelt csontrendszeri patológia van. A deformitás leggyakoribb típusa a tölcsér alakú deformitás. mellkas, kyphosis, scoliosis; ritkábban - a nyaki csigolyák számának csökkenése és fúziója, ami a Klippel-Feil szindróma anomáliáira emlékeztet.
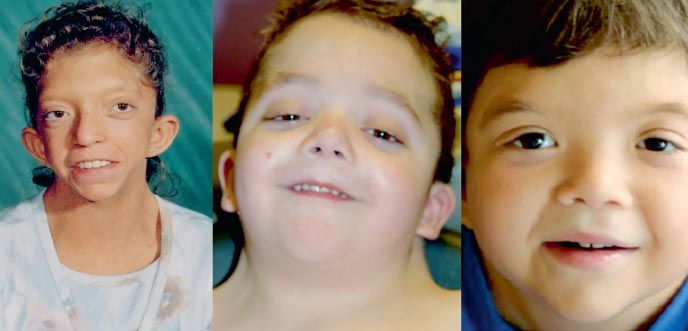
. A Noonan-szindróma fenotípusai
A Noonan-szindrómás betegeknek általában szőke, dús, göndör haja van, szokatlan növekedéssel a fej búbján; sötét foltok bőrön, hypertrichosis, körömlemezek disztrófiája, rendellenességek a fogak kitörésében és elhelyezkedésében, hajlamos keloid hegek kialakulására, fokozott bőr nyúlékonyság. A betegek egyharmadának perifériás nyiroködémája van, gyakrabban gyermekeknél fordul elő a kéz és a láb nyiroködémája fiatalon. Gyakori tünet a látás patológiája (myopia, strabismus, mérsékelt exophthalmus stb.). A növekedési retardáció a betegek körülbelül 75%-ánál fordul elő, fiúknál kifejezettebb és általában jelentéktelen. A növekedési retardáció az első életévekben nyilvánul meg, ritkábban enyhe születéskori magassági és súlyhiányok figyelhetők meg. Az élet első hónapjaitól kezdve az étvágy csökken. A csontkor általában elmarad az útlevél korától.
A szindróma jellegzetes vonása az unilaterális vagy kétoldali kriptorchidizmus, amely a férfi betegek 70-75%-ában fordul elő, felnőtteknél azoospermia, oligospermia, degeneratív változások herék. Ennek ellenére a pubertás spontán módon következik be, néha némi késéssel. A lányok gyakran tapasztalnak késést a menstruáció kialakulásában, és néha vannak szabálytalanságok menstruációs ciklus. A termékenység normális lehet mindkét nemnél.
Mentális retardációt a betegek több mint felénél észlelnek, általában kisebb. Viselkedési sajátosságokat, gátlástalanságot és figyelemhiányos zavart gyakran észlelnek. A beszéd általában jobban fejlett, mint mások intellektuális szférák. Az intelligencia hanyatlása nem korrelál a szomatikus rendellenességek súlyosságával [Marincheva G.S., 1988]. Elszigetelt esetekben a központi idegrendszer(hydrocephalus, spina bifida), thromboemboliás agyi infarktusok, amelyek esetleg vaszkuláris hypoplasiával társulnak.
Satu belső szervek a Noonan-szindrómára meglehetősen jellemző. A legjellemzőbbek a szív- és érrendszeri anomáliák: a pulmonalis artéria billentyűszűkülete (a betegek kb. 60%-a), hipertrófiás kardiomiopátia(20-30%), szerkezeti rendellenességek mitrális billentyű, pitvari sövény defektusok, Fallot tetralógiája; Az aorta koarktációját csak férfi betegeknél írták le.
A betegek egyharmadánál a húgyúti rendszer hibáit rögzítik (vese hypoplasia, medence duplikációja, hydronephrosis, megaureter stb.).
Elég gyakran Noonan-szindrómával fokozott vérzés figyelhető meg, különösen akkor, ha sebészeti beavatkozások V szájüregés nasopharynx. Különféle véralvadási hibákat észlelnek: a vérlemezke rendszer elégtelensége, a véralvadási faktorok, különösen a XI és XII szint csökkenése, megnövekedett thromboplastin idő. Beszámoltak a Noonan-szindróma leukémiával és rhabdomyosarcomával való kombinációjáról, ami a rosszindulatú daganatok kockázatának enyhe növekedését jelezheti ezeknél a betegeknél.
Az 1. táblázat a Noonan-szindróma fenotípusának jellemzőit mutatja be, amelyek a beteg életkorával változnak. A 2. táblázat a fenotípus és a genotípus közötti összefüggést mutatja Noonan-szindrómában.
Asztal 1. A Noonan-szindrómás betegek tipikus arcvonásai életkor szerint
| Homlok, arc, haj | Szemek | Fülek | Orr | Száj | Nyak | |
| Újszülött* | Magas homlok, alacsony hajvonal a fej hátsó részén | Hipertelorizmus, lefelé hajló palpebrális repedések, epicanthus redő | – | Rövid és széles süllyesztett gyökér, felfelé forduló hegy | Mélyen süllyesztett philtrum, az ajkak vörös határának magas, széles csúcsai, mikrognathia | Túlzott bőr a fej hátsó részén |
| Csecsemő (2-12 hónapos) | Nagy fej, magas és kiálló homlok | Hipertelorizmus, ptosis vagy vastagon lelógó szemhéjak | – | Rövid és széles süllyesztett gyökér | – | – |
| Gyermek (1-12 éves) | Durva vonások, hosszú arc | – | – | – | – | – |
| Tizenéves (12-18 éves) | Myopathiás arc | – | – | A híd magas és vékony | – | A nyaki ráncok nyilvánvaló kialakulása |
| Felnőtt (18 év felett) | A jellegzetes arcvonások kifinomultak, a bőr vékonynak és átlátszónak tűnik | – | – | Kiálló nasolabialis redő | – | – |
| Minden korosztály | – | Kék és zöld íriszek, rombusz alakú szemöldök | Alacsony, hátrafelé forgó fülek vastag redőkkel | – | – | – |
2. táblázat. A genotípus és a fenotípus közötti összefüggések Noonan-szindrómában*
| A szív- és érrendszer | Magasság | Fejlesztés | Bőr és haj | Egyéb | |
| PTPN11 (körülbelül 50%) | A tüdőtörzs szűkülete kifejezettebb; kevésbé – hipertrófiás kardiomiopátia és pitvari septum defektus | Rövidebb magasság; alacsonyabb IGF1 koncentráció | Az N308D-ben és N308S-ben szenvedő betegek intelligenciája enyhén csökkent vagy normális | – | A hemorrhagiás diathesis és a juvenilis myelomonocytás leukémia kifejezettebb |
| SOS1 (körülbelül 10%) | Kevesebb pitvari septum defektus | Magasabb növekedés | Kevesebb intelligenciacsökkenés, késleltetett beszédfejlődés | Hasonló a kardiokután-arc szindrómához | – |
| RAF1 (körülbelül 10%) | Súlyosabb hipertrófiás kardiomiopátia | – | – | Több anyajegyek, lentigo, café au lait foltok | – |
| KRAS (<2%) | – | – | Súlyosabb kognitív késés | Hasonló a kardiokután-arc szindrómához | – |
| NRAS (<1%) | – | – | – | – | – |
Laboratóriumi és funkcionális vizsgálatokból származó adatok
Nincsenek specifikus biokémiai markerek a Noonan-szindróma diagnosztizálására. Egyes betegeknél a növekedési hormon spontán éjszakai szekréciójának csökkenését észlelték a farmakológiai stimuláló tesztekre (klonidin és arginin) adott normális válasz mellett, a szomatomedin-C szintjének csökkenésével és a szomatomedinek válaszának csökkenésével növekedési hormon.
A diagnózis kritériumai
A Noonan-szindróma diagnózisa a klinikai tünetek alapján történik, egyes esetekben a diagnózist molekuláris genetikai vizsgálat eredményei igazolják. A szindróma diagnosztizálásának kritériumai közé tartozik a jellegzetes arc jelenléte (normál kariotípussal) a következő tünetek egyikével kombinálva: szívpatológia, alacsony termet vagy kriptorchidizmus (fiúkban), késleltetett pubertás (lányoknál). A kardiovaszkuláris patológia azonosításához a szív ultrahangvizsgálatát kell végezni az üregek méretének és a kamrák falának dinamikus meghatározásával. A betegség prenatális diagnózisa ultrahangos monitorozással lehetséges, amely lehetővé teszi a szívhibák és a nyak szerkezetének rendellenességeinek azonosítását.
Megkülönböztető diagnózis
Lányoknál a differenciáldiagnózis elsősorban Turner-szindróma; Egy citogenetikai vizsgálat tisztázza a diagnózist. A Noonan-szindróma fenotípusos jelei számos más betegségben is megtalálhatók: Williams-szindróma, LEOPARD-szindróma, Dubowitz-szindróma, cardiofaciocutan szindróma, Cornelia de Lange, Cohen, Rubinstein-Taybi stb. Ezeknek a betegségeknek a pontos azonosítása csak molekuláris vizsgálattal lesz lehetséges. az egyes szindrómák genetikai vizsgálata jelentős klinikai anyaggal, amely jelenleg aktív fejlesztés alatt áll.
Kezelés
A Noonan-szindrómás betegek kezelése a szív- és érrendszer hibáinak megszüntetésére, a mentális funkciók normalizálására, a növekedés és a szexuális fejlődés serkentésére irányul. A tüdőbillentyű diszpláziában szenvedő betegek kezelésére többek között a ballonos billentyűplasztikát is sikeresen alkalmazzák. A mentális fejlődés serkentésére nootróp és vaszkuláris gyógyszereket használnak. A szexuális fejlődést serkentő gyógyszerek főként kriptorchidizmusban szenvedő betegek számára javasoltak. A humán koriongonadotropin készítményeket életkor-specifikus dózisokban használják. Idősebb korban - hipogonadizmus jelenlétében - tesztoszteron készítmények. Az elmúlt években az emberi növekedési hormon rekombináns formáit használták a Noonan-szindrómás betegek kezelésére. A klinikai adatokat megerősíti a szomatomedin-C és a specifikus kötőfehérje szintjének növekedése a terápia során. A hosszú ideig növekedési hormon kezelésben részesülő betegek végső testmagassága esetenként meghaladja a családtagok átlagos testmagasságát.
Előrejelzés életre a szív- és érrendszeri patológia súlyossága határozza meg.
Megelőzés betegség orvosi genetikai tanácsadás adatain alapul.
Orvosi genetikai tanácsadás
Az orvosi genetikai tanácsadás lefolytatása során az autoszomális domináns öröklődési típusból és a betegség nagy (50%-os) kiújulásának kockázatából kell kiindulni az öröklött formájú családban. Az öröklődés típusának azonosítása érdekében alaposan meg kell vizsgálni a szülőket, mivel a szindróma minimális klinikai tünetekkel nyilvánulhat meg. Jelenleg a betegség molekuláris genetikai diagnosztikáját fejlesztették ki, és a következő gének mutációinak tipizálásával javítják: PTPN11, SOS1, RAF1, KRAS, NRAS stb. A betegség prenatális diagnosztizálására szolgáló módszerek fejlesztés alatt állnak.
Klinikai megfigyelés
A 9 éves G. fiút (3. kép) egy genetikus megfigyelte lakóhelyén „kromoszómapatológia?”, Williams-szindróma (sajátos fenotípus, a mitrális billentyűk leveleinek megvastagodása, hiperkalcémia 3 évente egyszer) diagnózisával. ?.
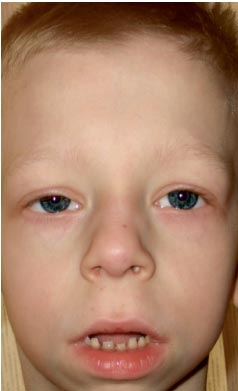
. A Noonan-szindrómás gyermek fenotípusának jellemzői (megnyúlt arccsont „pufók pofával”, rövid nyak, pterigoid ráncok a nyakon, rövidített orr előre nyitott orrlyukakkal, telt ajkak, ferde áll, anti-mongoloid bemetszés a palpebrális repedéseken , rossz elzáródás, macrostomia)
Panaszok csökkenti a memóriát, a fáradtságot, csökkenti a növekedési ütemet.
Családi történelem : a szülők nemzetiségüket tekintve oroszok, nem vér szerinti rokonok, és nincsenek foglalkozási veszélyeik, egészségesek. Az apa magassága 192 cm, az anya magassága 172 cm A törzskönyvben nem szerepelt mentális betegség, epilepszia, fejlődési elmaradás.
Élet- és betegségtörténet : egy fiú a 2. terhességből (1. terhesség - m/a), amely mindvégig vetélés veszélyével zajlott, polihidramnion kíséretében. Első szülés, időben, gyors, születési súly – 3400 g, hossza – 50 cm, azonnal felsikoltott, Apgar pontszáma – 7/9 pont. Születéskor a neonatológus felhívta a figyelmet a gyermek szokatlan fenotípusára és kariotípus vizsgálatot javasolt, az eredmény 46, XY (normál férfi kariotípus). Veleszületett pajzsmirigy alulműködésre gyanakodtak, pajzsmirigy profilt vizsgáltak, és az eredmény normális pajzsmirigy állapot lett. Ezután a gyermeket egy genetikus figyelte meg, feltételezhetően Williams-szindrómával. A korai posztnatális időszak jellemzők nélküli. Motorfejlődés életkor szerint, első szavak – egy év, frazális beszéd – 2 éves 3 hónapos korban.
8 éves korában endokrinológus konzultált vele a növekedési ütem csökkenése, a fáradtság és a memória romlása miatt. A kezek röntgenvizsgálata a csontkor (BA) mérsékelt elmaradását mutatta ki az útlevél életkorától (BA 6 évnek felelt meg). A pajzsmirigyprofil vizsgálata a pajzsmirigy-stimuláló hormon mérsékelt növekedését tárta fel, a szabad T4 normál szintjével és egyéb mutatókkal; A pajzsmirigy ultrahangja - patológia nélkül. Hormonterápiát írtak elő dinamikus megfigyeléssel.
Tekintettel a lakóhelyi diagnózis bizonytalanságára, a genetikus a gyermeket a Moszkvai Regionális Gyermekgyógyászati Konzultatív és Diagnosztikai Központba küldte a diagnózis tisztázása érdekében.
Objektív kutatási adatok:
Magasság - 126 cm, súly - 21 kg.
Fizikai fejlettsége átlag alatti, harmonikus. A növekedési Sds –1-nek felel meg (norma – –2+2). Fenotípus jellemzők (3. fotó): megnyúlt arcváz „pufók pofával”, rövid nyak, pterigoid redők a nyakon, alacsony szőrnövekedés a nyakon, rövidített orr előre nyitott orrlyukakkal, telt ajkak, ferde áll, anti-mongoloid bemetszés palpebrális repedések, macrostómia, mellbimbó-hipertelorizmus, mellkasi aszimmetria, a láb 2-3. ujjának hiányos bőrszindaktiliája, az interphalangealis ízületek súlyos hipermobilitása, törékeny, száraz körmök. Belső szervek – minden sajátosság nélkül. Szexuális fejlődés – Tanner I (amely a pubertás előtti időszaknak felel meg).
Laboratóriumi és funkcionális kutatási adatok:
A vér és a vizelet klinikai elemzése normális.
Biokémiai vérvizsgálat - a mutatók a normál határokon belül vannak.
Pajzsmirigy profil (TSH) – 7,5 µIU/ml (normál – 0,4–4,0), a többi indikátor normális.
Szomatotrop hormon (GH) – 7 ng/ml (normál – 7–10), somatomedin-C – 250 ng/ml (normál – 88–360).
A pajzsmirigy ultrahangja - patológia nélkül.
Belső szervek ultrahangja - minden jellemző nélkül.
EKG – sinus tachycardia, a szív elektromos tengelyének normál helyzete.
EchoCG - I. fokozatú MVP minimális regurgitációval, a mitrális billentyű szórólapjainak myxomatózus megvastagodása, egy további húr a bal kamra üregében.
A gerinc R-grafikája – a mellkasi gerinc jobb oldali scoliosisa, I. fokozat.
A kezek R-grafikája az alkar markolatával - csontkor 7-8 év.
Az epilepsziás aktivitás EEG-mintázatát nem rögzítették.
Az agy MRI-je - nincs kóros elváltozás.
Audiogram – patológia nélkül.
DNS-diagnosztika: molekuláris genetikai vizsgálat - a 7. kromoszóma kritikus régiójában nem észleltek deléciót a vizsgált lokuszokban; A Noonan-szindrómára jellemző Gly434Ary (1230G>A) mutációt az SOS1 gén 11. exonjában mutatták ki (a PTPN11 gén elemzése - mutációt nem találtunk).
Szakorvosi konzultációk:
Endokrinológus– szubklinikai hypothyreosis, hiányos gyógyszerkompenzáció.
Szemorvos- asztigmatizmus.
Neurológus– vegetatív-érrendszeri dystonia. Neurotikus reakciók.
Kardiológus- funkcionális kardiopátia.
Ortopéd sebészet- rossz testtartás. Mellkasi deformitás.
Genetikus- Noonan szindróma.
Figyelembe véve a gyermek fenotípusát, kórelőzményét és további vizsgálatok eredményeit, Noonan-szindróma diagnózist állítottak fel, amelyet egy molekuláris genetikai vizsgálat eredménye igazolt.
Így a bemutatott klinikai megfigyelés bemutatja a differenciáldiagnosztikai keresés nehézségeit, az egyedi jelek integrálásának szükségességét egy adott kóros állapot általános fenotípusába az örökletes betegségek egyedi formáinak célzott időben történő diagnosztizálása érdekében, valamint a molekuláris genetikai módszerek fontosságát a tisztázásban. a diagnózist. Az egyes szindrómák időben történő diagnosztizálása és genezisének tisztázása különösen fontos, mivel ezek lehetővé teszik az optimális megközelítés megtalálását ezen állapotok kezelésére és a lehetséges szövődmények megelőzésére (a gyermek fogyatékosságáig); örökletes betegségek kiújulásának megelőzése az érintett családokban (orvosi és genetikai tanácsadás). Ez azt diktálja, hogy a különböző szakterületek orvosai egyértelműen eligazodjanak az örökletes patológia áramlásában.
Bibliográfia:
- Baird P., De Jong B. Noonan-szindróma (XX és XY Turner fenotípus) egy család három generációjában // J. Pediatr., 1972, vol. 80. o. 110–114.
- Hasegawa T., Ogata T. et al. Az aorta és a vese hupoplázia koarktációja Turner/Noonan felszíni anomáliákkal és 46, XY kariotípussal rendelkező fiúban: klinikai modell egy feltételezett limfogén gén(ek) lehetséges károsodásához Turner szomatikus stigma esetén // Hum. Genet., 1996, vol. 97, r. 564–567.
- Fedotova T.V., Kadnikova V.A. et al. A Noonan-szindróma klinikai és molekuláris genetikai elemzése. Az Orosz Orvosi Genetikai Társaság VI. Kongresszusának anyagai. Orvosi genetika, 2010. 5. sz. melléklet, 184. o.
- Ward K.A., Moss C., McKeown C. A cardio-facio-cutan syndroma: a Noonan-szindróma megnyilvánulása? Br. J. Dermatol., 1994, vol. 131, r. 270–274.
- Municchi G., Pasquino A.M. et al. Növekedési hormon kezelés Noonan-szindrómában: jelentés négy olyan esetről, akik elérték a végső magasságot // Horm. Res., 1995, vol. 44, r. 164–167.
A gyermekeknél különféle veleszületett patológiák vannak. Néhányukat meglehetősen gyakorinak tekintik. De vannak ritka betegségek is. Elterjedtségük alacsony. Sokuknak azonban nagyon súlyos következményei vannak. Az egyik ilyen patológia a Noonan-szindróma. A következőkben nézzük meg részletesebben. A cikk leírja, hogyan nyilvánul meg a Noonan-szindróma, és miért jelenik meg. A szövegben egy fotót is bemutatunk a betegségről.
Általános információ
A Noonan-szindróma örökletes patológia. A mutáns PTPN11 gént az azt hordozó szülők utódjaihoz továbbítják. A férfiak általában terméketlenek. Ezért a gén az anyai vonalon keresztül jut tovább. Általában megjegyzik ennek a betegségnek a családi természetét. Ritka, nem öröklődő esetek, de a gyakorlatban is rögzítik őket.
Történelmi hivatkozás
Egy időben Jacqueline Noonan, aki gyermekkardiológusként dolgozott, miközben az Iowai Egyetem klinikáján dolgozott, észrevette, hogy a gyermekek egy bizonyos csoportját, amelyekben a tüdőartéria szűkületében szenvedő fiúk és lányok egyaránt tartalmaztak, gyakran alacsony termet jellemezte. tágra fektetett szemek és úszóhártyás nyakak, mélyen fekvő fülek és ptosis. Miután 833 betegen megvizsgálta a szívbetegség megnyilvánulásainak és más fejlődési rendellenességek kombinációját, az orvos 1962-ben írt egy cikket. Munkájában kilenc olyan esetet írt le, amikor a veleszületett típus hátterében jellegzetes arcvonások és alacsony termet mutatkozott. A betegséget férfiaknál és nőknél egyaránt észlelték. 
Noonan-szindróma: tünetek
Számos olyan jellemző van, amely megkülönbözteti a patológiát másoktól. Ide tartoznak különösen:
- Alacsony termetű. Nőknél - 1,53, férfiaknál - 1,63 m. Születéskor a gyermek testhossza és súlya is a normál határokon belül van. A növekedés 2-3 éves korban kezdődik.
- Az arc elváltozásai. A Noonan-szindrómával diagnosztizált betegek formái széles skálán mozognak. A belső sarokban bőrredő található. Ptosis (lelógó szemhéj) is megfigyelhető az izomműködés gyengülése vagy a kis szemüregek miatt. A patológiás gyermekek megközelítőleg kétharmada ptosisban szenved, és strabismus alakul ki.
- Zavarok az állkapcsok szerkezetében. A felső fejletlen, ívelt magas szájpadlás figyelhető meg. Mindkét állkapocsnál a fogak egymáshoz viszonyított helyzete nem megfelelő.
- Alacsony illeszkedés vagy a fül deformációja. Ennek az anomáliának a következtében a hallás romlik.
- Rövid és széles nyak.
- Pajzs alakú mellkas, szélesen elhelyezkedő mellbimbókkal.
- A könyökízület deformációja (veleszületett).
- Lúdtalp.
- Rövid ujjak.
A belső szervek rendellenességei
A Noonan-szindrómát leggyakrabban a szív- és érrendszer működésének zavarai kísérik. A patológia hátterében a tüdőtörzs szűkülete (stenosis), valamint az interventricularis septum hibája derül ki. A második helyet az urogenitális rendszer anomáliái foglalják el. Így a vesék részéről olyan hibák észlelhetők, mint a hipoplázia (a szövetfejlődés kudarca) vagy egy vese hiánya. Ami a pubertást illeti, ez a normáltól a teljesen hibásig változik. A lányoknál gyakran későn kezdődik a menstruáció, míg a fiúknak kriptorchidizmusuk van, vagy teljesen hiányoznak a herék. Spermatogenezis rendellenességeket is kimutatnak. Egyes esetekben a spermiumok teljesen hiányoznak. A Noonan-szindróma műtétei során fokozott vérzés figyelhető meg. Egyes betegeknél enyhe eltérések is előfordulhatnak. 
Űrlapok
Kétféle patológia létezik:
- Családi forma. Az autoszomális domináns típus örökletes jellege különbözteti meg. A mutáns gén hordozóinak patológiás utódai vannak.
- Szórványos forma. Ebben az esetben a mutáció esetről esetre megjelenik. Azonban nem azonosítottak örökletes tényezőt.
Okoz
A patológiát okozó leggyakoribb tényező a PTPN11 gén mutációja. Ezt az okot a betegek 50% -ánál észlelik. Az ebben a szindrómában szenvedő emberek bizonyos százalékánál azonban a genetikai tényező nem tisztázott. A patológia öröklődése autoszomális domináns formában fordul elő. A szindrómát egy új mutáció válthatja ki. Ebből az következik, hogy az ezzel a génnel rendelkező szülőknek nincs nagy esélyük arra, hogy újabb gyermeket szüljenek.
Diagnózis
A jellemző külső jelzéseknek megfelelően van felszerelve (ezeket fentebb ismertettük). A diagnózis során a laboratóriumi paramétereket is megvizsgálják. Különösen a tesztoszteron és a XII-es véralvadási faktor koncentrációja csökken. Műszeres vizsgálatokat is végeznek. A szegycsont röntgenfelvételét, az echokardiográfiát és az EKG-t írják elő. Ezekkel a módszerekkel a belső szervek anomáliáit észlelik. Az orvos konzultációt is előírhat 
Terápiás intézkedések
A szindróma genetikai természetéből adódóan a kezelés elsősorban a tünetek megszüntetésére irányul. A kriptorchidizmus esetén műtétet írnak elő. E folyamat során a herék a herezacskóba költöznek. Az androgénkoncentráció csökkenése hátterében hormonterápia javasolt. Veseelégtelenség esetén hemodialízist írnak elő. Segítségével az anyagcseretermékek extrarenális vértisztítással távoznak a szervezetből.
Noonan szindróma(J.A. Noonan, amerikai endokrinológus, született 1928-ban) egy örökletes betegség, amelyet alacsony termet és szomatikus fejlődési rendellenességek jellemeznek. Legteljesebben J. Noonen írta le 1963-ban. Lehet szórványos vagy családi, autoszomális recesszív módon öröklődő. Az N. s.-t okozó, kórosan megváltozott gének az autoszómákban lokalizálódnak (lásd. Örökletes betegségek ). Nemi kromatin és kariotípus (lásd. Kromoszómák ) megfelelnek a beteg nemének. A betegséget férfiaknál és nőknél egyaránt észlelik.
Az N. s. fő klinikai megnyilvánulásai. alacsony termet, alsó végtagok, szárny alakú bőrredők a nyakon, alacsony szőrnövekedési korlát a fej hátsó részén, jellegzetes dermatoglifikus elváltozások, hipertelorizmus, ptosis, anti-mongoloid szemforma, a fülszárnyak deformációja vagy alacsony helyzete , arc aszimmetria, széles orrnyereg, amitől a betegek hasonlítanak egymáshoz ( rizs .). Rövid nyak, veleszületett könyökízületi deformitás (cubitus valgus), brachydactylia, magas szájpadlás, kóros harapás , lapos lábak, a csigolyák alakjában és szerkezetében bekövetkezett változások és egyéb anomáliák Shereshevsky - Turner-szindróma . Gyakran észlelik a tüdő szűkületét és a kamrai sövény defektusát. A szív- és érrendszer egyéb veleszületett rendellenességei (lásd. Veleszületett szívhibák ) ritkábban figyelhetők meg. Szexuális fejlődés N. s. a normáltól a teljes ivarmirigy-dysgenesisig változik (lásd. Hermafroditizmus ). A lányoknál gyakran későn, míg a fiúknál gyakran későn kezdődik a menstruáció. kriptorchidizmus , hipogonadizmus . A herebiopsziából nyert anyag szövettani vizsgálata a csírasejtek csökkenését vagy teljes hiányát és a Leydig-sejtek hiperpláziáját mutatja. Az N. s. egyéb megnyilvánulásai. vannak gynecomastia és szellemi retardáció. A betegek vérszéruma tesztoszteront és ösztrogént tartalmaz (lásd. Nemi hormonok ) és gonadotropinok (lásd. Hipofízis hormonok ) normális, vagy az ivarmirigy-dysgenesis súlyosságától függően változik. A növekedési hormon koncentrációja a vérben normális. A 46XX vagy 46XV kariotípus megfelel az ivarmirigyek és az útlevél nemének.
A diagnózis a jellegzetes klinikai tünetek és laboratóriumi adatok alapján történik. N. s. leggyakrabban a Shereshevsky-Turner szindrómától különböztetik meg ( asztal .).
A hipogonadizmus tüneteinek kezelése nemi hormonokkal történő helyettesítésből áll. A növekedési hormon alkalmazása hatástalan. A javallatok szerint a veleszületett fejlődési rendellenességek műtéti korrekciója és a mentális retardációhoz kapcsolódó mentális zavarok kezelése történik (lásd. Endokrin mentális zavarok ). Az életre vonatkozó prognózis kedvező. Különös figyelmet kell fordítani a foglalkoztatásra: a szakválasztásnak hozzá kell járulnia a betegek szociális adaptációjához, figyelembe véve szellemi képességeiket és testi fogyatékosságukat.
A Noonen és Shereshevsky-Turner szindrómák differenciáldiagnosztikai jelei
Klinikai jel |
Noonan szindróma |
Shereshevsky-Turner szindróma |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Szexuális fejlődés |
A Noonan-szindrómát a Shereshevsky-Turner szindrómában szenvedő, normál kariotípusú betegekre jellemző többszörös veleszületett rendellenességek jellemzik. Néha a szindrómát Bonnevie-Ullrich szindrómával azonosítják, amely hasonló klinikai képpel rendelkezik. Bonnevie-Ullrich szindrómában azonban nincs szívelégtelenség, ami nagyon gyakran megfigyelhető Shereshevsky-Turner szindrómánál és Noonan. Minden betegnél kimutathatók a hipogonadizmus többé-kevésbé kifejezett tünetei, bár egyes szerzők úgy vélik, hogy a Noonan-szindróma esetén néhány fiúnál a nemi szervek normálisan fejlődnek. Etiológia. A szindróma dominánsan autoszomálisan öröklődik, alacsony génpenetranciával. A szindróma megoszlása meglehetősen nagy (1:1000 - 1:5000), de nincs egyértelmű diagnosztikai kritérium abnormális kariotípus formájában, mint a Shereshevsky-Turner-szindrómás betegeknél. Ennek eredményeként a betegség nem minden esetét észlelik. A hipotalamusz-hipofízis-ivarmirigy kapcsolatokat (patogenezist) Noonan-szindrómában gyakorlatilag nem vizsgálták. A páciens reproduktív rendszerének sajátosságairól egyetlen jelentés szól, amely alapján nehéz általánosítani. 8 Noonan-szindrómás fiúnál vizsgáltuk a tesztoszteron és a gonadotrop hormonok szintjét a vérben, valamint a herék funkcionális tartalékait. A tesztoszteronszint minden betegnél a normál alatt volt. A 17-KS vizelettel történő kiválasztása szintén csökkent. Ezek az adatok a herék hormonális hiányát jelzik Noonan-szindrómában. A tesztoszteron és a gonadotrop hormonok szintje a vérszérumban és a 17KC-kiválasztás a vizeletben (M±m) Noonan-szindrómás betegeknél
Az elégtelen tesztoszteron szekréció és a magas LH szint kombinációja 4 betegnél a herék, főként az intersticiális sejtek elsődleges elváltozását jelzi. A hCG 3 napon át 1500 E/m2 napi adagban történő alkalmazása azonban a tesztoszteronszint jelentős növekedéséhez vezetett ezeknek a fiúknak a vérében. Kauschunsky et al. (1977), amikor Noonan-szindrómás betegnek adták be a GnRH-t, a gonadotropinok szekréciójának kifejezett növekedését észlelték, mint az elsődleges hypogonadizmusban szenvedő betegeknél, és ezzel egyidejűleg a hCG-re adott pozitív herereakciót. A korlátozott megfigyelések megnehezítik az adatok értelmezését. Feltételezhető, hogy a betegeknél a Leydig-sejtek receptorainak érzékenysége az endogén gonadotropinra csökken, de a herék tartalékai megmaradnak, ami a tesztoszteron szekréció növekedésében tükröződik a nagy dózisú hCG-vel végzett stimuláció után. Alacsony tesztoszteronszinttel és a vérben normális gonadotrop hormonszinttel rendelkező betegeknél a hereelégtelenség patogenezise valószínűleg közel áll a normogonadotrop hipogonadizmus patogeneziséhez. Theintz és Savagl (1982) 5, 11,8-16,5 éves Noonan-szindrómában szenvedő fiút írtak le. Mindegyiküknél a hipogonadizmus egyértelmű jelei voltak, és késik a szexuális fejlődésük. Közülük 4 esetben a szexuális fejlődés normalizálódott, kettőnél spontán, kettőnél pedig tesztoszteron kezelés után. Csak egy betegnél volt hatástalan a hCG és a tesztoszteron kezelés. Így a Noonan-szindrómás betegeknél a közös klinikai kép ellenére a hipogonadizmus természete eltérő lehet. Reproduktív rendszerük elégtelensége az ivarmirigyek elsődleges hibájának, a hypothalamus-hipofízis rendszer működési zavarának és a herék gonadotrop stimulációval szembeni érzékenységének megváltozásának következménye lehet. A klinikai kép hasonlít a Shereshevsky-Turner-szindróma klinikájára. Kivonatot mutatunk be az egyik általunk megfigyelt beteg kórtörténetéből. A 8 éves Serezha L. a róla elnevezett kórház gyermekendokrinológiai osztályára került. Rauchfus 1982. március 5-én született, visszamaradt fizikai és szellemi fejlődéssel. Súlyos toxikózissal járó második terhességből született. A terhesség 7-8 hetében az anya akut légúti vírusfertőzést kapott. Az első terhesség vetéléssel végződött. Az anya veleszületett betegségben szenved. A fiú koraszülöttként született (császármetszéssel kinyerték). Testtömege 3500 g, hossza 51 cm Újszülött korban az állapot súlyos. 4 hónapos korban szívelégtelenséget diagnosztizáltak; a fiú már az első évtől lemaradt a testi és szellemi fejlődésben. A herék hiányát a herezacskóban születéskor észlelték. Felvételkor testhossza 124 cm (átlag alatti), súlya 22 kg. A testalkat rendellenes - széles, rövid nyak, aszimmetrikus mellkas, a szegycsont tölcsér alakú deformációja; magas szájpadlás, hipertelorizmus, füldeformáció, ujjhibák, szélesen elhelyezkedő mellbimbók. A pénisz kielégítően fejlett, a herezacskó nagyon kicsi, a herék nem tapinthatók. Az intelligencia mérsékelten csökken. Kariotípus 46XY. Röntgen a koponyáról minden jellemző nélkül. A csontok életkora 7 évnek felel meg. A vérszérumban az LH 4,33 mIU/ml, az FSH 3,13 mIU/ml, a tesztoszteron 0,56 nmol/l (minden mutató csökkent). A 17KS kiválasztódása 9,1 mmol/nap. Egyetlen hCG injekcióval végzett funkcionális teszt negatív volt - vér tesztoszteron - 0,57 nmol/l, 17-KS - 8,87 mmol/nap; A 3 napos teszt pozitív - tesztoszteron - 1,54 nmol/l, 17KS - 10,15 mmol/nap. Az anamnézis, a klinikai kép és a laboratóriumi adatok alapján Noonan-szindrómát diagnosztizáltak. A 3 napos pozitív teszt lehetővé tette a hCG kezelést. Ha nincs hatás, a cryptorchidizmus sebészeti kezelése javasolt. "Fiúk szexuális fejlődési zavarai" Kinézet.
Űrlapok
Okoz
DiagnosztikaA diagnózis a következők alapján történik:
Noonan-szindróma kezelése
Komplikációk és következmények
A Noonan-szindróma megelőzése
Hasonló cikkek
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||