Синдром на Нунан и подобни заболявания. Вижте какво е "синдром на Нунан" в други речници.
Е.В. Тозлиян, детски ендокринолог, генетик, кандидат на медицинските науки, Отделен структурно подразделение"Научноизследователски клиничен институт по педиатрия" SBEI VPO Руски национален изследователски медицински университет. Н.И. Пирогов на Министерството на здравеопазването на Руската федерация, Москва Ключови думиКлючови думи: деца, синдром на Нунан, диагностика.
ключови думи: деца, синдром на Нунан, диагностика.
Статията описва синдрома на Нунан (синдром на Улрих-Нунан, тернероиден синдром с нормален кариотип) - рядък вродена патология, унаследено по автозомно-доминантен начин, е семеен характерСрещат се обаче и спорадични случаи. Синдромът предполага наличието на фенотип, характерен за синдрома на Шерешевски-Търнър при индивиди от женски и мъжки пол с нормален кариотип. Представено клинично наблюдение. Сложността на диференциално диагностичното търсене, липсата на информираност на клиницистите за този синдроми значението на интердисциплинарния подход.
Исторически факти
За първи път около необичаен синдромспоменава О. Кобилински през 1883 г. (снимка 1).
най-старият известен клиничен случайСиндром на Нунан, описан през 1883 г. от О. Кобилински
Заболяването е описано през 1963 г. от американския кардиолог Жаклин Нунан, която съобщава за девет пациенти с клапна стеноза. белодробна артериянисък ръст, хипертелоризъм, умерен интелектуален спад, птоза, крипторхизъм и скелетни нарушения. Д-р Нунан, който практикува като детски кардиологв Университета на Айова, забеляза, че децата с рядък типсърдечно заболяване - стеноза на белодробната клапа - често се наблюдават типични физически аномалии под формата на нисък ръст, криловидна врата, широко поставени очи и ниско поставени уши. Момчетата и момичетата бяха еднакво възхитени. Д-р Джон Опиц, бивш ученикНунан е първият, който въвежда термина "синдром на Нунан", за да опише състоянието на деца, които имат признаци, подобни на тези, описани от Нунан. По-късно Нунан написа статията "Хипертелоризъм с фенотипа на Търнър", а през 1971 г. името "синдром на Нунан" беше официално признато на симпозиума по сърдечно-съдови заболявания.
Етиология и патогенеза
Синдромът на Noonan е автозомно доминантно разстройство с променлива експресивност (фиг. 1). Генът на синдрома на Нунан е локализиран в дълго рамохромозома 12. Генетичната хетерогенност на синдрома не може да бъде изключена. Описани са спорадични и фамилни форми на синдрома с автозомно-доминантна форма на наследяване. При семейни случаи мутантен геннаследени, като правило, от майката, тъй като поради тежки малформации пикочно-половата системамъжете с това състояние често са безплодни. Повечето от докладваните случаи са спорадични, причинени от de novo мутации.
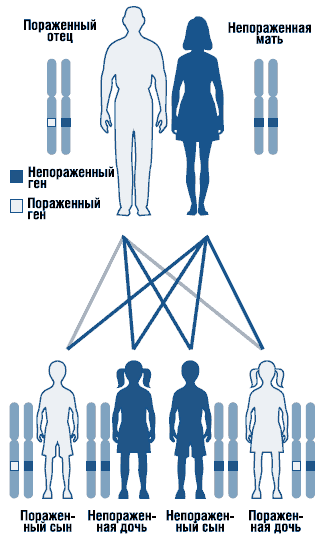
. Автозомно доминантен модел на наследяване
Описаните комбинации от синдром на Нунан с неврофиброматоза тип I в няколко семейства предполагат възможна връзка между два независими локуса 17q11.2 на хромозома 17. Някои пациенти имат микроделеции в локуса 22q11 на хромозома 22; в тези случаи клинични проявленияСиндромите на Noonan се комбинират с хипотиреоидизъм на тимуса и синдром на DiGeorge. Редица автори обсъждат участието на предполагаеми гени на лимфогенезата в патогенезата на синдрома поради наличието на лицеви и соматични аномалии, подобни на синдрома на Turner, и висока честота на патология. лимфна система.
Повечето обща каузаСиндромът на Noonan е мутация на гена PTPN11, която се открива при приблизително 50% от пациентите. Протеинът, кодиран от гена PTPN11, принадлежи към семейство молекули, които регулират реакцията на еукариотните клетки към външни сигнали. Най-голямо числомутациите в синдрома на Noonan са локализирани в екзони 3, 7 и 13 на гена PTPN11, кодиращи протеинови домени, отговорни за прехода на протеина към активно състояние.
Възможните идеи за патогенезата са представени от следните механизми:
Пътят на RAS-MAPK е много важен път на сигнална трансдукция, през който преминават извънклетъчните лиганди определени факторирастеж, цитокини и хормони – стимулират клетъчната пролиферация, диференциация, оцеляване и метаболизъм (фиг. 2). След свързване на лиганда, рецепторите на клетъчната повърхност се фосфорилират в местата на тяхната ендоплазмена област. Това свързване включва адапторни протеини (напр. GRB2), които образуват конститутивен комплекс с гуанин нуклеотидни обменни фактори (напр. SOS), които превръщат неактивния GDP-свързан RAS в неговата активна GTP-свързана форма. След това активираните RAS протеини активират каскадата RAF-MEKERK чрез серия от реакции на фосфорилиране. В резултат на това активираният ERK навлиза в ядрото, за да промени транскрипцията на целевите гени и коригира активността на ендоплазмените цели, за да предизвика адекватни краткосрочни и дългосрочни клетъчни отговори на стимула. Всички гени, участващи в синдрома на Noonan, кодират протеини, неразделна част от този път, и мутации причиняващи болести, обикновено усилват сигнала, преминаващ през този път.
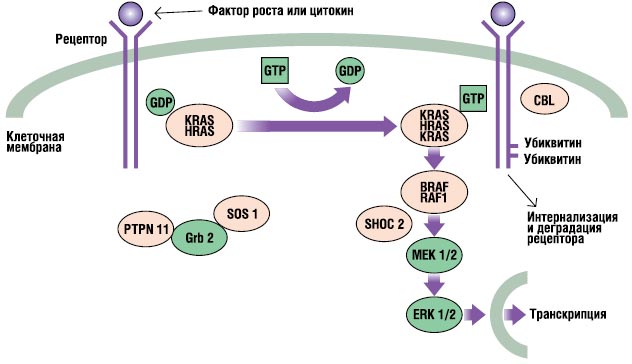
. RAS-MAPK сигнален път. Сигналите за растеж се предават с активирани рецептори на растежен фактор към ядрото. Мутациите в PTPN11, KRAS, SOS1, NRAS и RAF1 са свързани със синдрома на Noonan, а мутациите в SHOC2 и CBL са свързани с фенотип, подобен на синдрома на Noonan.
Клинични характеристики на синдрома на Noonan
Фенотипът на пациенти със синдром на Нунан прилича на синдрома на Търнър: къс вратс птеригоидна гънка или нисък растеж на косата, нисък ръст, хипертелоризъм на палпебралните фисури (снимка 2). Лицевите микроаномалии включват антимонголоиден разрез на палпебралните фисури, латерален кантус надолу, птоза, епикантус, ниско разположени ушни миди, сгъната къдрица ушни миди, неправилно захапване, цепнатина на увулата меко небце, готическо небе, микрогнатия и микрогения. Гръдният кош на щитовидната жлеза е с хипопластични широко разположени зърна, гръдната кост е изпъкнала в горната част и потъва в долната. Около 20% от пациентите имат умерено тежка патология на скелета. Най-честата деформация на фунията гръден кош, кифоза, сколиоза; по-рядко - намаляване на броя на шийните прешлени и тяхното сливане, наподобяващо аномалии при синдрома на Klippel-Feil.
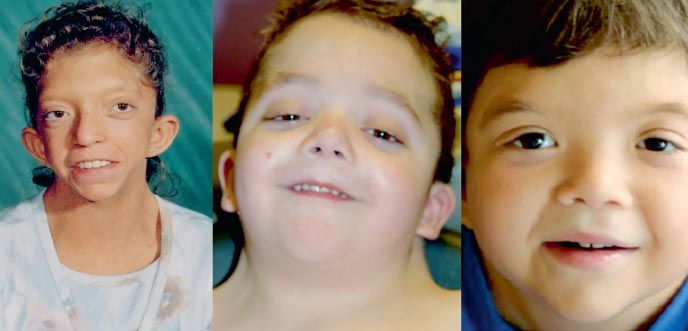
. Фенотипове на синдрома на Noonan
Пациентите със синдром на Нунан обикновено имат руса, гъста, къдрава коса с необичаен растеж на темето, често тъмни петнавърху кожата, хипертрихоза, дегенерация на нокътните плочки, аномалии в никненето и разположението на зъбите, склонност към образуване на келоидни белези, повишена разтегливост на кожата. Една трета от пациентите имат периферен лимфедем, по-често лимфедем на ръцете и краката се проявява при деца ранна възраст. Чест признак е патология на зрението (миопия, страбизъм, умерен екзофталм и др.). Забавянето на растежа се среща при приблизително 75% от пациентите, по-изразено е при момчетата и обикновено е незначително. Забавянето на растежа се проявява през първите години от живота, по-рядко има лек дефицит в растежа и теглото при раждането. От първите месеци на живота се наблюдава намаляване на апетита. Костната възраст обикновено изостава от паспортната.
Характерна особеност на синдрома е едностранният или двустранен крипторхизъм, който се среща при 70-75% от пациентите от мъжки пол; при възрастни пациенти, азооспермия, олигоспермия, дегенеративни променитестисите. Независимо от това пубертетът настъпва спонтанно, понякога с известно закъснение. При момичетата често има забавяне на образуването на менструация, понякога - нарушения менструален цикъл. Фертилитетът може да е нормален и при двата пола.
Умствена изостаналост се открива при повече от половината пациенти, обикновено незначителна. Често се отбелязват поведенчески особености, дезинхибиция, разстройство с дефицит на вниманието. Речта обикновено е по-добре развита от другите интелектуални сфери. Степента на намаляване на интелигентността не корелира с тежестта на соматичните разстройства [Marincheva G.S., 1988]. В изолирани случаи малформации на централната нервна система(хидроцефалия, гръбначни хернии), тромбоемболични инфаркти на мозъка, вероятно свързани със съдова хипоплазия.
пороци вътрешни органисъс синдрома на Noonan са доста характерни. Най-характерни са сърдечно-съдовите аномалии: клапна стеноза на белодробната артерия (около 60% от пациентите), хипертрофична кардиомиопатия(20–30%), структурни аномалии митрална клапа, дефекти на междупредсърдната преграда, тетралогия на Fallot; коарктация на аортата е описана само при пациенти от мъжки пол.
При една трета от пациентите се регистрират малформации на пикочната система (хипоплазия на бъбреците, удвояване на таза, хидронефроза, мегауретер и др.).
Доста често при синдром на Noonan се наблюдава повишено кървене, особено при хирургични интервенции V устната кухинаи назофаринкса. Откриват се различни коагулационни дефекти: недостатъчност на тромбоцитната система, намаляване на нивото на коагулационните фактори, особено XI и XII, увеличаване на тромбопластиновото време. Има съобщения за комбинация от синдром на Noonan с левкемия и рабдомиосаркома, което може да показва леко повишаване на риска от злокачествено заболяване при тези пациенти.
Таблица 1 представя характеристиките на фенотипа при синдрома на Noonan, които се променят с възрастта на пациента. Таблица 2 показва корелацията между фенотипа и генотипа при синдрома на Noonan.
маса 1. Типични черти на лицето при пациенти със синдром на Noonan по възраст
| Чело, лице, коса | очи | Уши | нос | Устата | Шия | |
| новородено* | Високо чело, ниска линия на косата в тилната област | Хипертелоризъм, наклонен надолу палпебрални фисури, епикантална гънка | – | Къс и широк вдлъбнат корен, обърнат нагоре връх | Дълбоко вдлъбнат филтрум, високи широки върхове на червената граница на устните, микрогнатия | Излишна кожа на гърба на главата |
| Гърди (2–12 месеца) | Голяма глава, високо и изпъкнало чело | Хипертелоризъм, птоза или дебели увиснали клепачи | – | Къс и широк вдлъбнат корен | – | – |
| Дете (1-12 години) | Груби черти, дълго лице | – | – | – | – | – |
| Тийнейджър (12-18 години) | Миопатично лице | – | – | Мостът е висок и тънък | – | Явно образуване на гънки на врата |
| Възрастен (>18 години) | Отличителните черти на лицето са изтънчени, кожата изглежда тънка и полупрозрачна | – | – | Изпъкнала назолабиална гънка | – | – |
| Всички възрасти | – | Сини и зелени ириси, вежди във формата на диамант | Ниски, завъртяни назад уши с плътни гънки | – | – | – |
таблица 2. Корелации между генотип и фенотип при синдром на Noonan*
| Сърдечно-съдовата система | Височина | развитие | Кожа и коса | други | |
| PTPN11 (прибл. 50%) | Стенозата на белодробния ствол е по-изразена; по-малко - хипертрофична кардиомиопатия и дефект на предсърдната преграда | По-нисък растеж; по-ниска концентрация на IGF1 | Пациентите с N308D и N308S имат лек спад или нормален интелект | – | По-изразена хеморагична диатеза и ювенилна миеломоноцитна левкемия |
| SOS1 (прибл. 10%) | По-малък дефект на предсърдната преграда | По-висок растеж | По-малко намаляване на интелигентността, забавено развитие на речта | Подобно на кардиокутанния лицев синдром | – |
| RAF1 (прибл. 10%) | По-тежка хипертрофична кардиомиопатия | – | – | | Повече ▼ родилни петна, лентиго, петна от кафе с мляко | – |
| КРАС (<2%) | – | – | По-тежко когнитивно забавяне | Подобно на кардио-кожно-лицевия синдром | – |
| NRAS (<1%) | – | – | – | – | – |
Данни от лабораторни и функционални изследвания
Няма специфични биохимични маркери за диагностициране на синдрома на Нунан. При някои пациенти се наблюдава намаляване на спонтанната нощна секреция на растежен хормон с нормален отговор на тестове за фармакологична стимулация (клофелин и аргинин), намаляване на нивото на соматомедин-С и намаляване на отговора на соматомедините към въвеждането на растежен хормон. са открити.
Критерии за диагностика
Диагнозата "синдром на Noonan" се поставя въз основа на клинични признаци, в някои случаи диагнозата се потвърждава от резултатите от молекулярно-генетично изследване. Критериите за диагностициране на синдрома включват наличието на характерно лице (с нормален кариотип) в комбинация с един от следните признаци: сърдечно заболяване, нисък ръст или крипторхизъм (при момчета), забавен пубертет (при момичета). За откриване на сърдечно-съдова патология е необходимо да се проведе ултразвуково изследване на сърцето с динамично определяне на размера на кухините и стените на вентрикулите. Пренаталната диагностика на заболяването е възможна с помощта на ултразвуково наблюдение, което позволява да се открият сърдечни дефекти и аномалии в структурата на шията.
Диференциална диагноза
При момичетата диференциалната диагноза се прави предимно със синдрома на Търнър; Диагнозата може да се изясни чрез цитогенетично изследване. Фенотипните признаци на синдрома на Noonan се откриват при редица други заболявания: синдром на Уилямс, синдром на LEOPARD, Dubovitz, сърдечно-лицево-кожен синдром, Cornelia de Lange, Cohen, Rubinstein-Taybi и др. Точното идентифициране на тези заболявания ще бъде възможно само при провеждане на молекулярно-генетични изследвания на всеки синдром със значителен клиничен материал, който в момента се разработва активно.
Лечение
Лечението на пациенти със синдром на Noonan е насочено към премахване на дефекти на сърдечно-съдовата система, нормализиране на умствените функции, стимулиране на растежа и сексуалното развитие. За лечение на пациенти с дисплазия на клапите на белодробната артерия, наред с други методи, успешно се използва балонна валвулопластика. За да се стимулира умственото развитие, се използват ноотропни и съдови средства. Лекарствата, насочени към стимулиране на сексуалното развитие, са показани главно при пациенти с крипторхизъм. Препаратите с хорион гонадотропин се използват във възрастови дози. В по-напреднала възраст - при наличие на хипогонадизъм - тестостеронови препарати. През последните години рекомбинантни форми на човешки растежен хормон се използват при лечението на пациенти със синдром на Noonan. Клиничните данни се потвърждават от повишаване на нивото на соматомедин-С и специфичния свързващ протеин по време на терапията. Крайният ръст на пациентите, получаващи дългосрочна терапия с растежен хормон, в някои случаи надвишава средния ръст на членовете на семейството.
Прогноза за цял живот се определя от тежестта на сърдечно-съдовата патология.
Предотвратяване заболяване се основава на данни от медицинско генетично консултиране.
Медицинско генетично консултиране
При медицинско генетично консултиране трябва да се изхожда от автозомно-доминантния тип наследство и висок (50%) риск от рецидив на заболяването в семейството с наследствени форми. За да се идентифицира естеството на вида на наследството, е необходимо да се извърши задълбочен преглед на родителите, тъй като синдромът може да се прояви с минимални клинични симптоми. В момента е разработена и се усъвършенства молекулярно-генетична диагностика на заболяването чрез типизиране на мутации в гените: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разработват се методи за пренатална диагностика на заболяването.
Клинично наблюдение
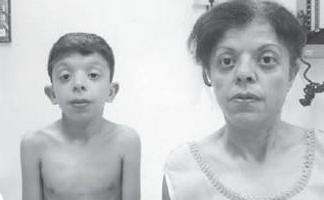
Момче Г., 9 години (снимка 3), е наблюдавано по местоживеене от генетик с диагноза хромозомна патология?, синдром на Уилямс (особен фенотип, удебеляване на куспидите на митралната клапа, хиперкалциемия веднъж на всеки 3 години) ?.
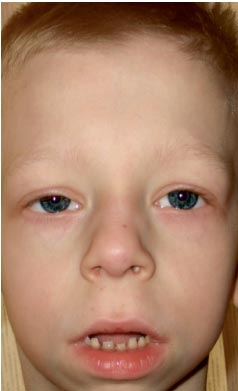
. Особености на фенотипа на дете със синдром на Noonan (удължен лицев скелет с „пухкави бузи“, къс врат, криловидни гънки на шията, скъсен нос с отворени напред ноздри, подпухнали устни, наклонена брадичка, антимонголоид инцизия на палпебралните фисури, неправилна оклузия, макростомия)
Оплаквания върху намалена памет, умора, намалени темпове на растеж.
Семейна история : родителите са руски по националност, нямат кръвна връзка и нямат професионални рискове, здрави. Височината на бащата е 192 см, височината на майката е 172 см. В родословието на случаи на психични заболявания, епилепсия, забавяне на развитието не са отбелязани.
История на живота и болестта : момче от 2-ра бременност (1-ва бременност - m / a), която продължи със заплаха от прекъсване през цялото време, придружена от полихидрамнион. Раждането беше първо, навреме, бързо, тегло - 3400 гр., дължина - 50 см. Изпищя веднага, оценка по Апгар - 7/9 точки. При раждането неонатологът обърна внимание на необичайния фенотип на детето, препоръча изследването на кариотипа, резултатът е 46, XY (нормален мъжки кариотип). Съмнение за вроден хипотиреоидизъм, направено е изследване на тиреоиден профил, резултатът е нормален тиреоиден статус. Освен това детето е наблюдавано от генетик с предполагаема диагноза "синдром на Уилямс". Ранен постнатален период - без особености. Моторно развитие според възрастта, първите думи - до годината, фразовата реч - на 2 години и 3 месеца.
На 8 години е консултиран от ендокринолог за намален растеж, умора и намалена памет. Рентгеновото изследване на ръцете показва умерено изоставане в костната възраст (BC) от паспортната (BC съответства на 6 години). Проучването на профила на щитовидната жлеза разкрива умерено увеличение на тиреостимулиращия хормон с нормално ниво на свободен Т4 и други показатели; Ехография на щитовидна жлеза - без патология. Предписана е хормонална терапия, последвана от динамично наблюдение.
Като се има предвид несигурността на диагнозата по местоживеене, генетикът насочва детето към Московския регионален консултативен и диагностичен център за деца, за да изясни диагнозата.
Данни от обективно изследване:
Височина - 126 см, тегло - 21 кг.
Физическото развитие е под средното, хармонично. Sds на растежа съответства на -1 (нормално -2 + 2). Характеристики на фенотипа (снимка 3): удължен лицев скелет с "пухкави бузи", къс врат, птеригоидни гънки на шията, нисък растеж на косата на шията, къс нос с отворени напред ноздри, подпухнали устни, наклонена брадичка, антимонголоиден инцизия на палпебралните фисури, малоклузия, макростомия, хипертелоризъм на зърната, асиметрия на гръдния кош, непълна кожна синдактилия на 2-ри или 3-ти пръст на краката, изразена хипермобилност на интерфалангеалните стави, чупливи, сухи нокти. По вътрешните органи - без особености. Сексуално развитие - Танер I (което съответства на предпубертетния период).
Данни от лабораторни и функционални изследвания:
Клиничният анализ на кръвта и урината е норма.
Биохимичен анализ на кръвта - показатели в рамките на нормата.
Профил на щитовидната жлеза (TSH) - 7,5 μIU / ml (норма - 0,4-4,0), други показатели са нормални.
Соматотропен хормон (STH) - 7 ng / ml (норма - 7-10), соматомедин-C - 250 ng / ml (норма - 88-360).
Ехография на щитовидна жлеза - без патология.
Ехография на вътрешни органи - без особености.
ЕКГ - синусова тахикардия, нормалното положение на електрическата ос на сърцето.
EchoCG - MVP от 1-ва степен с минимална регургитация, миксоматозно удебеляване на куспидите на митралната клапа, допълнителна хорда в кухината на лявата камера.
R-графия на гръбначния стълб - дясностранна сколиоза на гръдния отдел на гръбначния стълб I степен.
R-графия на ръцете с улавяне на предмишниците - костна възраст 7–8 години.
ЕЕГ модели на епилептична активност не са регистрирани.
ЯМР на мозъка - без патологични изменения.
Аудиограма - без патология.
ДНК диагностика: молекулярно-генетично изследване - не са открити делеции на изследваните локуси на критичната област на хромозома 7; Установена е мутация Gly434Ary (1230G>A) в 11-ия екзон на гена SOS1 (анализ на ген PTPN11 - не са открити мутации), което е типично за синдрома на Noonan.
Експертен съвет:
Ендокринолог- субклиничен хипотиреоидизъм, непълна лекарствена компенсация.
Оптометрист- астигматизъм.
Невролог- вегетативна дистония. невротични реакции.
Кардиолог- функционална кардиопатия.
Хирург-ортопед- нарушение на позата. Деформация на гръдния кош.
ГенетикСиндром на Нунан.
Като се вземат предвид фенотипа на детето, данните от историята, резултатите от допълнителни изследвания, беше поставена диагнозата синдром на Noonan, която беше потвърдена от резултата от молекулярно-генетично изследване.
По този начин представеното клинично наблюдение показва сложността на диференциално диагностичното търсене, необходимостта от интегриране на отделни признаци в общия фенотип на конкретно патологично състояние за целенасочена навременна диагностика на определени форми на наследствени заболявания и значението на молекулярно-генетичните методи за изясняване на диагноза. Навременната диагноза, изясняването на генезиса на всеки синдром са особено важни, тъй като ви позволяват да намерите най-добрия подход към лечението на тези състояния, предотвратяване на възможни усложнения (до инвалидност на детето); предотвратяване на повторна поява на наследствени заболявания в засегнатите семейства (медико-генетично консултиране). Това налага необходимостта лекарите от различни специалности ясно да се ориентират в потока на наследствената патология.
Библиография:
- Baird P., De Jong B. Синдром на Noonan (XX и XY фенотип на Turner) в три поколения на семейство // J. Pediatr., 1972, том. 80, стр. 110–114.
- Хасегава Т., Огата Т. и др. Коарктация на аортата и бъбречна хупоплазия при момче с повърхностни аномалии на Turner/Noonan и кариотип 46, XY: клиничен модел за възможно увреждане на предполагаем лимфогенен ген(и) за соматични стигмати на Turner // Hum. Генет., 1996, кн. 97, стр. 564–567.
- Федотова Т.В., Кадникова В.А. et al. Клинико-молекулярно-генетичен анализ на синдрома на Нунан. Материали от VI конгрес на Руското дружество по медицинска генетика. Медицинска генетика, Приложение към бр.5, 2010, стр.184.
- Ward K.A., Moss C., McKeown C. Кардио-фацио-кутанен синдром: проява на синдрома на Noonan? // Br. J. Dermatol., 1994, том. 131, стр. 270–274.
- Municchi G., Pasquino A.M. et al. Лечение с хормон на растежа при синдром на Noonan: доклад за четири случая, които са достигнали крайна височина // Horm. Res., 1995, том. 44, стр. 164–167.
При децата има различни вродени патологии. Някои от тях се считат за доста често срещани. Но има и редки болести. Тяхното разпространение е ниско. Много от тях обаче имат доста сериозни последствия. Една от тези патологии е синдромът на Noonan. След това го разгледайте по-подробно. Статията ще опише как и защо се появява синдромът на Noonan. В текста ще бъде представена и снимка на заболяването.
Главна информация
Синдромът на Noonan е наследствена патология. Мутиралият ген PTPN11 от родителите, които го носят, се предава на потомството. По правило мъжете са безплодни. Следователно генът се предава по майчина линия. Обикновено се отбелязва фамилният характер на това заболяване. Редки случаи, които не са свързани с наследствеността, обаче, те също са регистрирани в практиката.
Историческа справка
По едно време Жаклин Нунан, която практикува като педиатричен кардиолог, докато работи в клиника в Университета на Айова, забелязва, че определена група деца, която включва както момчета, така и момичета с белодробна стеноза, често се различават по нисък ръст, широко отворени очи, ципести вратове, ниско разположени ушни миди и птоза. След като изследва комбинацията от прояви на сърдечни заболявания с други аномалии в развитието при 833 пациенти, лекарят написа статия през 1962 г. В работата си тя описва девет случая, при които са отбелязани характерни черти на лицето и нисък ръст на фона на вроден тип. Заболяването се открива както при мъже, така и при жени. 
Синдром на Нунан: симптоми
Има редица характерни черти, които отличават патологията от другите. Те включват по-специално:
- Нисък ръст. При жените - 1,53, при мъжете - 1,63 м. Към момента на раждане както дължината на тялото, така и теглото на детето са в рамките на нормалното. Забавянето на растежа започва на 2-3 години.
- Промени в лицето. Пациентите, диагностицирани със синдром на Noonan, имат широко разположени форми. Във вътрешния ъгъл има кожна гънка. Има и птоза (увиснали клепачи) поради отслабена мускулна функция или поради малки очни кухини. Приблизително две трети от всички деца с патология имат страбизъм.Когато птозата е ограничена и се развива страбизъм.
- Нарушения на структурата на челюстите. Горната е недоразвита, има извито високо небце. И в двете челюсти позицията на зъбите е неправилна.
- Ниско кацане или деформация на ушите. В резултат на тази аномалия слухът е нарушен.
- Къса и широка шия.
- Щит гръдния кош с широко разположени зърна.
- Деформация в лакътната става (вродена).
- Плоски стъпала.
- Къси пръсти.
Малформации на вътрешните органи
Синдромът на Noonan най-често се придружава от нарушения в дейността на сърдечно-съдовата система. На фона на патологията се открива стесняване (стеноза) в белодробния ствол, както и дефект в интервентрикуларната преграда. Второ място заемат аномалиите на пикочно-половата система. Така че от страна на бъбреците се разкриват дефекти като хипоплазия (липса на развитие на тъкан) или липса на един бъбрек. Що се отнася до пубертета, той варира от нормален до напълно дефектен. Момичетата често имат късно начало на менструацията, момчетата имат крипторхизъм или тестисите напълно отсъстват. Има и нарушения на сперматогенезата. В някои случаи сперматозоидите напълно отсъстват. На фона на операции със синдром на Noonan се отбелязва повишено кървене. Някои пациенти също могат да покажат леки аномалии. 
Форми
Има два вида патология:
- семейна форма. Различава се в наследствения характер на автозомно-доминантния тип. При носителите на мутантния ген потомството се появява с патология.
- спорадична форма. В този случай мутацията се появява от случай на случай. В този случай наследственият фактор не е идентифициран.
причини
Най-честият фактор, провокиращ патологията, е мутация в гена PTPN11. Тази причина се открива при 50% от пациентите. При определен процент от хората с този синдром обаче генетичният фактор е неясен. Наследяването на патологията протича в автозомно-доминантна форма. Синдромът може да бъде предизвикан от нова мутация. От това следва, че родителите с такъв ген нямат голям шанс да имат още едно дете.
Диагноза
Монтира се в съответствие с характерните външни характеристики (те са описани по-горе). При диагностиката се изследват и лабораторни показатели. По-специално, има намаляване на концентрацията на тестостерон, коагулационен фактор XII. Провеждат се и инструментални изследвания. Предписва се рентгенова снимка на гръдната кост, ехокардиография, ЕКГ. С помощта на тези методи се откриват аномалии на вътрешните органи. Лекарят може също да назначи консултация 
Терапевтични дейности
Поради генетичното състояние на синдрома, лечението е насочено главно към елиминиране на симптомите. При крипторхизъм се предписва операция. По време на него тестисите се преместват в скротума. На фона на намаляване на концентрацията на андрогени се препоръчва хормонална терапия. При наличие на бъбречна недостатъчност се предписва хемодиализа. С негова помощ продуктите от метаболитните процеси се отстраняват от тялото чрез извънбъбречно пречистване на кръвта.
Синдром на Нунан(J.A. Noonan, американски ендокринолог, роден през 1928 г.) е наследствено заболяване, характеризиращо се с нисък ръст и аномалии в соматичното развитие. J. Nunen е описан най-пълно през 1963 г. Може да бъде спорадичен или фамилен, унаследяван по автозомно-рецесивен начин. Патологично променените гени, причиняващи Н. с., са локализирани в автозоми (вж. наследствени заболявания ). Полов хроматин и кариотип Хромозоми ) отговарят на пола на пациента. Заболяването се среща както при мъжете, така и при жените.
Основните клинични прояви на Н. с. нисък ръст, долни крайници, птеригоидни гънки на кожата на шията, ниска линия на косата в задната част на главата, характерни дерматоглифни промени, хипертелоризъм, птоза, антимонголоиден разрез на очите, деформация или ниско прилягане на ръбовете на ушите, асиметрия на лице, широк мост на носа, което прави пациентите подобни един на друг приятел ( ориз .). Характеризира се с къс врат, вродена деформация на лакътната става (cubitus valgus), брахидактилия, високо небце, патологични хапя , плоски стъпала, промени във формата и структурата на прешлените и други аномалии, отбелязани по време на Синдром на Шерешевски-Търнър . Често се открива стеноза на белодробния ствол, дефект на камерната преграда. Други вродени малформации на сърдечно-съдовата система (вж. Вродени сърдечни дефекти ) са по-рядко срещани. Сексуално развитие при Н. стр. варира от нормална до пълна гонадна дисгенезия (вж Хермафродитизъм ). Момичетата често имат късно начало на менструацията, момчетата - крипторхизъм , хипогонадизъм . Хистологичното изследване на материала, получен от тестикуларна биопсия, показва намаляване или пълно отсъствие на зародишни клетки и хиперплазия на клетките на Leydig. Други прояви на Н. с. са гинекомастия и умствена изостаналост. В кръвния серум на пациенти съдържанието на тестостерон, естрогени (вж. полови хормони ) и гонадотропини (вж хормони на хипофизата ) е нормално или променено в зависимост от тежестта на гонадалната дисгенезия. Концентрацията на растежен хормон в кръвта е нормална. Кариотип 46XX или 46XV съответства на гонаден и паспортен пол.
Диагнозата се поставя въз основа на характерни клинични признаци и лабораторни данни. Н. с. най-често се диференцира със синдрома на Шерешевски-Търнър ( маса .).
Лечението при наличие на признаци на хипогонадизъм се състои в заместителна терапия с препарати от полови хормони. Използването на растежен хормон е неефективно. Според показанията, хирургична корекция на вродени малформации, лечение на психични разстройства, свързани с умствена изостаналост (вж. Ендокринни психични разстройства ). Прогнозата за живота е благоприятна. Особено внимание трябва да се обърне на заетостта: изборът на специалност трябва да допринесе за социалната адаптация на пациентите, като се вземат предвид техните умствени способности и физически увреждания.
Диференциално-диагностични характеристики на синдромите на Noonan и Shereshevsky-Turner
клиничен признак |
Синдром на Нунан |
Синдром на Шерешевски-Търнър |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
половото развитие |
Синдромът на Нунан се характеризира с множество вродени малформации, типични за пациенти със синдром на Шерешевски-Търнър, с нормален кариотип. Понякога синдромът се идентифицира със синдрома на Bonnevie-Ulrich, който има подобна клинична картина. Въпреки това, при синдрома на Bonnevie-Ulrich няма сърдечно заболяване, което много често се наблюдава при синдрома на Shereshevsky-Turner и Нунан. При всички пациенти могат да се открият повече или по-малко изразени симптоми на хипогонадизъм, въпреки че някои автори смятат, че при синдрома на Noonan гениталиите се развиват нормално при някои момчета. Етиология. Синдромът се унаследява по автозомно-доминантен начин с ниска генна пенетрантност. Разпределението на синдрома е доста голямо (1: 1000 - 1: 5000), но няма ясен диагностичен критерий под формата на анормален кариотип, както при пациенти със синдром на Шерешевски-Търнър. В тази връзка не всички случаи на заболяването се откриват. Хипоталамо-хипофизно-половите връзки (патогенеза) при синдрома на Noonan практически не са проучени. Има само един доклад за особеностите на репродуктивната система при пациент, въз основа на който е трудно да се правят обобщения. Изследвахме съдържанието на тестостерон и гонадотропни хормони в кръвта, както и функционалните резерви на тестисите при 8 момчета със синдром на Noonan. Нивата на тестостерон при всички пациенти са под нормата. Екскрецията на 17-KS в урината също е намалена. Тези открития сочат тестикуларен хормонален дефицит при синдрома на Noonan. Серумни нива на тестостерон и гонадотропни хормони и уринна екскреция на 17KS (M±m) при пациенти със синдром на Noonan
Комбинацията от недостатъчна секреция на тестостерон и високи нива на LH при 4 пациенти показва първична лезия на тестисите, главно интерстициални клетки. Въпреки това, приложението на CG в продължение на 3 дни в дневна доза от 1500 IU/m2 доведе до значително повишаване на нивата на тестостерон в кръвта при тези момчета. Kauschunsky и др. (1977), когато прилагат GnRH на пациент със синдром на Noonan, те отбелязват изразено повишаване на секрецията на гонадотропини, както при пациенти с първичен хипогонадизъм, и в същото време положителна реакция на тестисите към CG. Ограниченият брой наблюдения затруднява тълкуването на данните. Може да се предположи, че при пациентите чувствителността на рецепторите на клетките на Leydig към ендогенния гонадотропин е намалена, но резервите на тестисите са запазени, което се изразява в повишаване на секрецията на тестостерон след стимулация с високи дози hCG. При пациенти с ниски нива на тестостерон и нормални нива на гонадотропни хормони в кръвта, патогенезата на тестикуларната недостатъчност вероятно е близка до патогенезата на нормогонадотропния хипогонадизъм. Theintz, Savagl (1982) описва 5 момчета на възраст 11,8 - 16,5 години със синдром на Noonan. Всички са с ясни признаци на хипогонадизъм и изоставане в половото развитие. При 4 от тях след това половото развитие се нормализира, като при двама спонтанно, а при двама - след лечение с тестостерон. Само при един пациент лечението с CG и тестостерон е неефективно. По този начин при пациенти със синдром на Noonan, въпреки сходството на клиничната картина, естеството на хипогонадизма може да бъде различно. Недостатъчността на репродуктивната система при тях може да бъде резултат от първичен дефект в половите жлези, дисфункция на хипоталамо-хипофизната система и промени в чувствителността на тестисите към гонадотропна стимулация. Клиничната картина наподобява клиниката на синдрома на Шерешевски-Търнър. Ето извадка от историята на един от пациентите, които наблюдавахме. Сережа Л., на 8 години, е приет в детското ендокринологично отделение на болницата. Rauhfus на 03/05/82 с изоставане във физическото и умственото развитие. Той е роден от втората бременност, която протича с тежка токсикоза. В 7-8 седмица от бременността майката е прекарала остра респираторна вирусна инфекция. Първата бременност завърши със спонтанен аборт. Майката страда от вродена Момчето е родено преждевременно (извлечено с цезарово сечение). Телесно тегло 3500 гр., дължина 51 см. В неонаталния период състоянието е тежко. На 4-месечна възраст е диагностицирано сърдечно заболяване; момчето изостана във физическото и умственото развитие от първата година. Липсата на тестиси в скротума е открита при раждането. При постъпване дължина на тялото 124 см (под средното), тегло 22 кг. Телосложението е неправилно - широк къс врат, асиметричен гръден кош, фуниевидна деформация на гръдната кост; високо небце, хипертелоризъм, деформация на ухото, дефекти на пръстите, широко разположени зърна. Пенисът е развит задоволително, скротумът е много малък, тестисите не се напипват. Интелигентността е умерено намалена. Кариотип 46XY. Рентгенова снимка на черепа без особености. Костната възраст съответства на 7 години. В кръвния серум LH 4,33 mIU / ml, FSH 3,13 mIU / ml, тестостерон 0,56 nmol / l (всички показатели са намалени). Екскреция 17KS 9,1 mmol/ден. Функционален тест с еднократно инжектиране на hCG е отрицателен - кръвен тестостерон - 0,57 nmol/l, 17-KS - 8,87 mmol/ден; 3-дневният тест е положителен - тестостерон - 1,54 nmol / l, 17KS - 10,15 mmol / ден. Въз основа на анамнезата, клиничната картина и лабораторните данни е поставена диагноза синдром на Noonan. Положителният 3-дневен тест позволи лечение на CG. При липса на ефект е показано хирургично лечение на крипторхизъм. "Нарушения на половото развитие при момчета", Външен вид.
Форми
причини
ДиагностикаДиагнозата се установява въз основа на:
Лечение на синдрома на Noonan
Усложнения и последствия
Профилактика на синдрома на Noonan
Подобни статии
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||