Noonans syndrom och liknande sjukdomar. Se vad "Noonan Syndrome" är i andra ordböcker.
E.V. Tozliyan, pediatrisk endokrinolog, genetiker, kandidat för medicinska vetenskaper, Separat strukturell indelning"Scientific Research Clinical Institute of Pediatrics" SBEI VPO Russian National Research Medical University. N.I. Pirogov från Ryska federationens hälsoministerium, Moskva Nyckelord Nyckelord: barn, Noonans syndrom, diagnostik.
nyckelord: barn, Noonans syndrom, diagnostik.
Artikeln beskriver Noonans syndrom (Ulrich-Noonans syndrom, terneroid syndrom med normal karyotyp) - en sällsynt medfödd patologi, ärvt på ett autosomalt dominant sätt, är familjekaraktär Men sporadiska fall förekommer också. Syndromet antyder närvaron av en fenotyp som är karakteristisk för Shereshevsky-Turners syndrom hos kvinnliga och manliga individer med en normal karyotyp. Presenteras klinisk observation. Komplexiteten i differentialdiagnostiksökning, bristande medvetenhet hos läkare om detta syndrom och vikten av ett tvärvetenskapligt förhållningssätt.
Historiska fakta
För första gången ungefär ovanligt syndrom omtalad av O. Kobylinski 1883 (foto 1).
äldsta kända kliniskt fall Noonans syndrom, beskrivet 1883 av O. Kobylinski
Sjukdomen beskrevs 1963 av den amerikanska kardiologen Jacqueline Noonan, som rapporterade om nio patienter med klaffstenos. lungartären, kortväxthet, hypertelorism, måttlig intellektuell nedgång, ptos, kryptorchidism och skelettsjukdomar. Dr Noonan, som praktiserade som barnkardiolog vid University of Iowa, märkte att barn med sällsynt typ hjärtsjukdom - stenos i lungklaffen - ofta observerade typiska fysiska anomalier i form av kortväxthet, pterygoid nacke, breda ögon och lågt liggande öron. Pojkar och flickor var lika förvånade. Dr John Opitz, före detta student Noonan, var den första som introducerade termen "Noonans syndrom" för att beskriva tillståndet hos barn som hade tecken som liknade de som beskrevs av Noonan. Senare skrev Noonan artikeln "Hypertelorism with the Turner phenotype", och 1971 erkändes namnet "Noonans syndrom" officiellt vid symposiet för hjärt- och kärlsjukdomar.
Etiologi och patogenes
Noonans syndrom är en autosomal dominant störning med varierande uttrycksförmåga (Fig. 1). Noonans syndrom genen är lokaliserad till lång axel kromosom 12. Den genetiska heterogeniteten hos syndromet kan inte uteslutas. Sporadiska och familjära former av syndromet med en autosomal dominant form av nedärvning har beskrivits. I familjefall mutant genärvt, som regel, från modern, eftersom på grund av svåra missbildningar genitourinary systemet män med detta tillstånd är ofta infertila. De flesta av de rapporterade fallen är sporadiska, orsakade av de novo-mutationer.
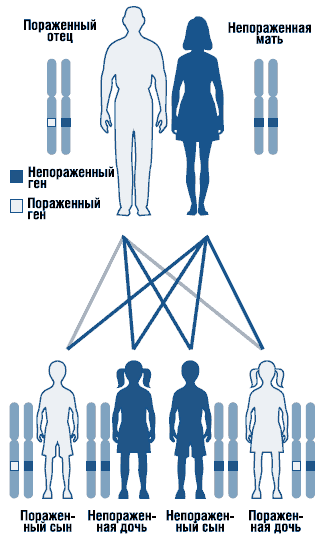
. Autosomalt dominant arvsmönster
De beskrivna kombinationerna av Noonans syndrom med typ I neurofibromatos i flera familjer antydde ett möjligt samband mellan två oberoende loci 17q11.2 av kromosom 17. Vissa patienter har mikrodeletioner i 22q11 locus av kromosom 22; i dessa fall kliniska manifestationer Noonans syndrom kombineras med hypotyreos av tymus och DiGeorges syndrom. Ett antal författare diskuterar involveringen av förmodade gener av lymfogenes i patogenesen av syndromet på grund av närvaron av ansikts- och somatiska anomalier liknande Turners syndrom och en hög förekomst av patologi. lymfsystemet.
Mest vanlig orsak Noonans syndrom är en mutation av PTPN11-genen, som finns hos cirka 50 % av patienterna. Proteinet som kodas av PTPN11-genen tillhör en familj av molekyler som reglerar eukaryota cellers svar på externa signaler. Största antalet mutationer i Noonans syndrom är lokaliserade i exonerna 3, 7 och 13 av PTPN11-genen, som kodar för proteindomäner som är ansvariga för övergången av proteinet till det aktiva tillståndet.
Möjliga idéer om patogenes representeras av följande mekanismer:
RAS-MAPK-vägen är en mycket viktig signaltransduktionsväg genom vilken extracellulära ligander är vissa faktorer tillväxt, cytokiner och hormoner - stimulerar cellproliferation, differentiering, överlevnad och metabolism (Fig. 2). Efter ligandbindning fosforyleras receptorer på cellytan vid platserna för deras endoplasmatiska region. Denna bindning involverar adapterproteiner (t.ex. GRB2) som bildar ett konstitutivt komplex med guanin-nukleotidutbytesfaktorer (t.ex. SOS) som omvandlar det inaktiva GDP-bundna RAS till dess aktiva GTP-bundna form. De aktiverade RAS-proteinerna aktiverar sedan RAF-MEKERK-kaskaden genom en serie fosforyleringsreaktioner. Som ett resultat kommer aktiverad ERK in i kärnan för att ändra transkriptionen av målgener och korrigerar aktiviteten hos endoplasmatiska mål för att inducera adekvata kortsiktiga och långvariga cellulära svar på stimulansen. Alla gener involverade i Noonans syndrom kodar för proteiner som är integrerade i denna väg, och mutationer sjukdomsframkallande, förstärker vanligtvis signalen som passerar genom denna väg.
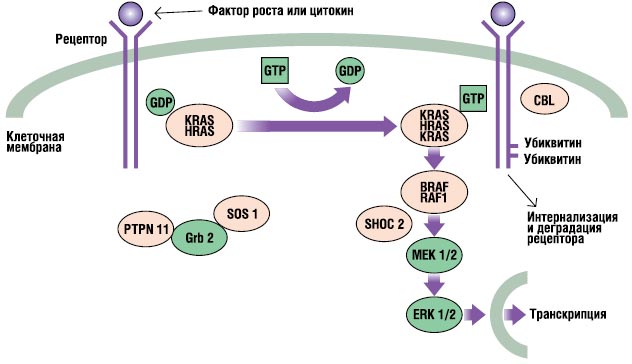
. RAS-MAPK signalväg. Tillväxtsignaler överförs med aktiverade tillväxtfaktorreceptorer till kärnan. Mutationer i PTPN11, KRAS, SOS1, NRAS och RAF1 är associerade med Noonan syndrom, och mutationer i SHOC2 och CBL är associerade med en Noonan syndrom-liknande fenotyp.
Kliniska egenskaper hos Noonans syndrom
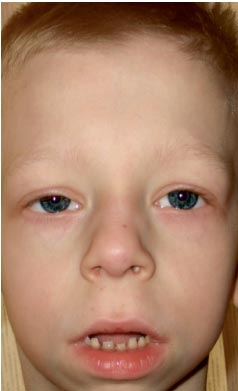
Fenotypen hos patienter med Noonans syndrom liknar Turners syndrom: kort hals med ett pterygoidveck eller låg hårväxt, kortväxthet, hypertelorism av palpebrala fissurer (foto 2). Mikroanomalier i ansiktet inkluderar antimongoloid snitt i palpebralfissurerna, nedåtriktad lateral canthus, ptosis, epicanthus, lågt liggande öronbågar, vikt krullning auriklar, malocklusion, kluven uvula mjuk gom, gotisk himmel, mikrognati och mikrogeni. Sköldkörtelns bröstkorg bildas med hypoplastiska breda bröstvårtor, bröstbenet sticker ut i den övre delen och sjunker i den nedre. Cirka 20% av patienterna har måttligt svår patologi i skelettet. Den vanligaste trattdeformiteten bröst kyfos, skolios; mindre ofta - en minskning av antalet halskotor och deras fusion, som liknar anomalier i Klippel-Feil syndrom.
. Fenotyper av Noonans syndrom
Patienter med Noonans syndrom har vanligtvis blont, tjockt, lockigt hår med ovanlig krontillväxt, ofta mörka fläckar på huden, hypertrichosis, degeneration av nagelplattorna, anomalier i utbrott och arrangemang av tänder, en tendens att bilda keloidärr, ökad hudtöjbarhet. En tredjedel av patienterna har perifert lymfödem, oftare manifesteras lymfödem i händer och fötter hos barn tidig ålder. Ett frekvent tecken är synpatologi (närsynthet, skelning, måttlig exophthalmos, etc.). Tillväxtretardation förekommer hos cirka 75 % av patienterna, är mer uttalad hos pojkar och är vanligtvis obetydlig. Tillväxthämning visar sig under de första levnadsåren, mer sällan finns det ett litet underskott i tillväxt och vikt vid födseln. Från de första månaderna av livet sker en minskning av aptiten. Benåldern släpar vanligtvis efter passåldern.
Ett karakteristiskt kännetecken för syndromet är unilateral eller bilateral kryptorkism, som förekommer hos 70–75% av manliga patienter; hos vuxna patienter, azoospermi, oligospermi, degenerativa förändringar testiklar. Ändå uppstår puberteten spontant, ibland med viss fördröjning. Hos flickor finns det ofta en försening i bildandet av menstruation, ibland - kränkningar menstruationscykel. Fertiliteten kan vara normal hos båda könen.
Mental retardation upptäcks hos mer än hälften av patienterna, vanligtvis mindre. Beteendeegenskaper, disinhibition, uppmärksamhetsstörning noteras ofta. Tal är vanligtvis bättre utvecklat än andra intellektuella sfärer. Graden av minskning av intelligens korrelerar inte med svårighetsgraden av somatiska störningar [Marincheva G.S., 1988]. I enstaka fall, missbildningar av den centrala nervsystem(hydrocefalus, ryggmärgsbråck), tromboemboliska infarkter i hjärnan, möjligen associerade med vaskulär hypoplasi.
laster inre organ med Noonans syndrom är ganska karakteristiska. De mest typiska är kardiovaskulära anomalier: valvulär stenos i lungartären (cirka 60% av patienterna), hypertrofisk kardiomyopati(20–30%), strukturella anomalier mitralisklaffen, förmaksseptumdefekter, tetralogi av Fallot; coarctation av aorta har beskrivits endast hos manliga patienter.
Hos en tredjedel av patienterna registreras missbildningar i urinvägarna (hypoplasi i njurarna, fördubbling av bäckenet, hydronefros, megaureter, etc.).
Ganska ofta, med Noonans syndrom, noteras ökad blödning, särskilt med kirurgiska ingrepp V munhålan och nasofarynx. Olika koagulationsdefekter hittas: insufficiens av trombocytsystemet, en minskning av nivån av koagulationsfaktorer, särskilt XI och XII, en ökning av tromboplastintiden. Det finns rapporter om en kombination av Noonans syndrom med leukemi och rabdomyosarkom, vilket kan tyda på en lätt ökning av risken för malignitet hos dessa patienter.
Tabell 1 visar egenskaperna hos fenotypen vid Noonans syndrom, som förändras med patientens ålder. Tabell 2 visar korrelationen mellan fenotyp och genotyp vid Noonans syndrom.
bord 1. Typiska ansiktsdrag hos patienter med Noonans syndrom efter ålder
| Panna, ansikte, hår | Ögon | Öron | Näsa | Mun | Nacke | |
| Nyfödd* | Hög panna, låg hårfäste i occipitalområdet | Hypertelorism, nedåtlutande palpebrala sprickor, epikantal veck | – | Kort och bred försänkt rot, uppåtvänd spets | Djupt försänkt philtrum, höga breda toppar av läpparnas vermiljonkant, micrognathia | Överskott av hud på baksidan av huvudet |
| Bröst (2–12 månader) | Stort huvud, högt och utstående panna | Hypertelorism, ptos eller tjocka hängande ögonlock | – | Kort och bred infälld rot | – | – |
| Barn (1-12 år) | Grova drag, långt ansikte | – | – | – | – | – |
| Tonåring (12-18 år) | Myopatiskt ansikte | – | – | Bron är hög och smal | – | Uppenbar nackveckbildning |
| Vuxen (>18 år) | Distinkta ansiktsdrag är förfinade, huden ser tunn och genomskinlig ut | – | – | Utskjutande nasolabialveck | – | – |
| Alla åldrar | – | Blå och gröna iris, diamantformade ögonbryn | Låga, bakåtroterade öron med tjocka veck | – | – | – |
Tabell 2. Korrelationer mellan genotyp och fenotyp vid Noonans syndrom*
| Det kardiovaskulära systemet | Höjd | Utveckling | Hud och hår | Övrig | |
| PTPN11 (cirka 50 %) | Stenosen av en lungstam är mer uttryckt; mindre - hypertrofisk kardiomyopati och förmaksseptumdefekt | Lägre tillväxt; lägre koncentration av IGF1 | Patienter med N308D och N308S har mild minskning eller normal intelligens | – | Mer uttalad hemorragisk diatese och juvenil myelomonocytisk leukemi |
| SOS1 (ca 10 %) | Mindre förmaksseptumdefekt | Högre tillväxt | Mindre nedgång i intelligens, försenad talutveckling | Liknar kardiokutant ansiktssyndrom | – |
| RAF1 (ca 10 %) | Svårare hypertrofisk kardiomyopati | – | – | Mer födelsemärken, lentigo, fläckar av kaffe med mjölk | – |
| KRAS (<2%) | – | – | Mer allvarlig kognitiv försening | Liknar kardio-kutant-ansiktssyndrom | – |
| NRAS (<1%) | – | – | – | – | – |
Data från laboratorie- och funktionsstudier
Det finns inga specifika biokemiska markörer för diagnosen Noonans syndrom. Hos vissa patienter, en minskning av spontan nattlig utsöndring av tillväxthormon med ett normalt svar på farmakologiska stimuleringstester (klofelin och arginin), en minskning av nivån av somatomedin-C och en minskning av svaret av somatomediner på införandet av tillväxthormon upptäcks.
Diagnoskriterier
Diagnosen "Noonan syndrom" görs på grundval av kliniska tecken, i vissa fall bekräftas diagnosen av resultaten av en molekylärgenetisk studie. Kriterier för att diagnostisera syndromet inkluderar närvaron av ett karakteristiskt ansikte (med en normal karyotyp) i kombination med en av följande egenskaper: hjärtsjukdom, kortväxthet eller kryptorkism (hos pojkar), försenad pubertet (hos flickor). För att upptäcka kardiovaskulär patologi är det nödvändigt att utföra en ultraljudsundersökning av hjärtat med en dynamisk bestämning av storleken på hålrummen och ventriklarnas väggar. Prenatal diagnos av sjukdomen är möjlig med hjälp av ultraljudsövervakning, vilket gör det möjligt att upptäcka hjärtfel och anomalier i nackens struktur.
Differentialdiagnos
Hos flickor ställs differentialdiagnosen i första hand med Turners syndrom; Diagnosen kan klargöras genom cytogenetisk undersökning. Fenotypiska tecken på Noonans syndrom finns i ett antal andra sjukdomar: Williams syndrom, LEOPARD syndrom, Dubovitz, kardiofacio-kutant syndrom, Cornelia de Lange, Cohen, Rubinstein-Taybi, etc. Noggrann identifiering av dessa sjukdomar kommer endast att vara möjlig när man genomför molekylärgenetiska studier av varje syndrom med betydande kliniskt material som för närvarande aktivt utvecklas.
Behandling
Behandling av patienter med Noonans syndrom syftar till att eliminera defekter i det kardiovaskulära systemet, normalisera mentala funktioner, stimulera tillväxt och sexuell utveckling. För behandling av patienter med dysplasi av klaffarna i lungartären, bland andra metoder, används ballongvalvuloplastik framgångsrikt. För att stimulera mental utveckling används nootropa och vaskulära medel. Läkemedel som syftar till att stimulera sexuell utveckling är främst indicerade för patienter med kryptorkism. Koriongonadotropinpreparat används i åldersdoser. Vid en högre ålder - i närvaro av hypogonadism - testosteronpreparat. Under senare år har rekombinanta former av humant tillväxthormon använts vid behandling av patienter med Noonans syndrom. Kliniska data bekräftas av en ökning av nivån av somatomedin-C och specifikt bindande protein under behandlingen. Den slutliga längden för patienter som får långtidsbehandling med tillväxthormon överstiger i vissa fall medellängden för familjemedlemmar.
Prognos för livet bestäms av svårighetsgraden av kardiovaskulär patologi.
Förebyggande sjukdomen är baserad på data från medicinsk genetisk rådgivning.
Medicinsk genetisk rådgivning
Inom medicinsk genetisk rådgivning bör man utgå från den autosomalt dominanta typen av ärftlighet och hög (50 %) risk för återfall av sjukdomen i familjen med ärftliga former. För att identifiera arten av typen av arv är det nödvändigt att genomföra en grundlig undersökning av föräldrarna, eftersom syndromet kan manifestera sig med minimala kliniska symtom. För närvarande har molekylärgenetisk diagnos av sjukdomen utvecklats och förbättras genom typning av mutationer i generna: PTPN11, SOS1, RAF1, KRAS, NRAS, etc. Metoder för prenatal diagnos av sjukdomen utvecklas.
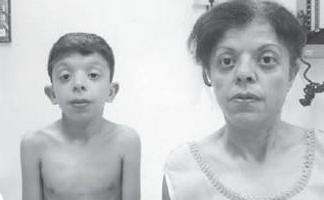
Klinisk observation
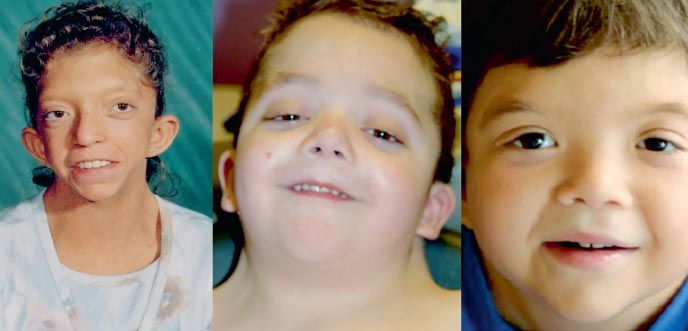
Pojken G., 9 år gammal (foto 3), observerades på bostadsorten av en genetiker med diagnosen kromosomal patologi?, Williams syndrom (en märklig fenotyp, förtjockning av mitralisklaffen, hyperkalcemi en gång vart tredje år) ?.
. Egenskaper hos fenotypen hos ett barn med Noonans syndrom (ett långsträckt ansiktsskelett med "knubbiga kinder", en kort hals, pterygoida veck på nacken, en förkortad näsa med näsborrarna öppna framåt, svullna läppar, en sluttande haka, en anti-mongoloid snitt av palpebrala fissurer, malocklusion, makrostomi)
Klagomål på minskat minne, trötthet, minskad tillväxthastighet.
Familjehistoria : föräldrar är ryska till nationalitet, inte släkt med blod och inte har yrkesmässiga risker, friska. Faderns höjd är 192 cm, moderns höjd är 172 cm. I stamtavlan för fall av psykisk sjukdom, epilepsi, noterades inte utvecklingsförseningar.
Historia om liv och sjukdom : en pojke från 2:a graviditeten (1:a graviditeten - m / a), som fortsatte med hot om avbrott hela tiden, åtföljd av polyhydramnios. Förlossningen var den första, i tid, snabb, födelsevikt - 3400 g, längd - 50 cm. Han skrek direkt, Apgar-poäng - 7/9 poäng. Vid födseln uppmärksammade neonatologen den ovanliga fenotypen av barnet, rekommenderade studien av karyotypen, resultatet är 46, XY (normal manlig karyotyp). Medfödd hypotyreos misstänktes, en sköldkörtelprofilstudie gjordes, resultatet var ett normalt sköldkörtelstatus. Vidare observerades barnet av en genetiker med en förmodad diagnos av "Williams syndrom". Tidig postnatal period - utan drag. Motorisk utveckling efter ålder, de första orden - efter år, frasalt tal - vid 2 år 3 månader.
Vid 8 års ålder rådfrågades han av en endokrinolog om minskad tillväxthastighet, trötthet och minskat minne. En röntgenundersökning av händerna visade en måttlig eftersläpning i benålder (BC) från pass ett (BC motsvarade 6 år). Studien av sköldkörtelprofilen visade en måttlig ökning av sköldkörtelstimulerande hormon med en normal nivå av fritt T4 och andra indikatorer; Ultraljud av sköldkörteln - utan patologi. Hormonbehandling ordinerades, följt av dynamisk observation.
Med hänsyn till osäkerheten i diagnosen på bostadsorten hänvisade genetikern barnet till Moskvas regionala rådgivande och diagnostiska centrum för barn för att klargöra diagnosen.
Objektiv forskningsdata:
Höjd - 126 cm, vikt - 21 kg.
Den fysiska utvecklingen är under genomsnittet, harmonisk. Tillväxt Sds motsvarar -1 (normal -2 + 2). Funktioner hos fenotypen (foto 3): långsträckt ansiktsskelett med "knubbiga kinder", kort hals, pterygoida veck på nacken, låg hårväxt på nacken, kort näsa med näsborrarna öppna framåt, svullna läppar, sluttande haka, anti-mongoloid snitt av palpebrala fissurer, malocklusion , makrostomi, hypertelorism i bröstvårtan, asymmetri i bröstet, ofullständig hudsyndaktyli av 2:a eller 3:e fingrarna på fötterna, uttalad överrörlighet i interfalangeala lederna, sköra, torra naglar. På de inre organen - utan funktioner. Sexuell utveckling - Tanner I (som motsvarar den prepubertala perioden).
Data från laboratorie- och funktionsstudier:
Klinisk analys av blod och urin är normen.
Biokemisk analys av blod - indikatorer inom normalområdet.
Sköldkörtelprofil (TSH) - 7,5 μIU / ml (normal - 0,4-4,0), andra indikatorer är normala.
Somatotropt hormon (STH) - 7 ng / ml (normalt - 7-10), somatomedin-C - 250 ng / ml (normalt - 88-360).
Ultraljud av sköldkörteln - utan patologi.
Ultraljud av de inre organen - utan funktioner.
EKG - sinustakykardi, den normala positionen för hjärtats elektriska axel.
EchoCG - MVP av 1: a graden med minimal uppstötning, myxomatös förtjockning av mitralisklaffens cusps, en extra korda i håligheten i vänster kammare.
R-grafi av ryggraden - högersidig skolios i bröstryggen, I grad.
R-grafi av händerna med fångst av underarmarna - benålder 7–8 år.
EEG-mönster av epileptisk aktivitet registrerades inte.
MRI av hjärnan - utan patologiska förändringar.
Audiogram - utan patologi.
DNA-diagnostik: molekylärgenetisk studie - inga deletioner av de studerade loci av den kritiska regionen av kromosom 7 upptäcktes; Gly434Ary (1230G>A)-mutation hittades i den 11:e exonen av SOS1-genen (PTPN11-genanalys - inga mutationer hittades), vilket är typiskt för Noonans syndrom.
Expertråd:
Endokrinolog- subklinisk hypotyreos, ofullständig läkemedelskompensation.
Optiker- astigmatism.
Neurolog- vegetativ dystoni. neurotiska reaktioner.
Kardiolog- funktionell kardiopati.
Ortopedisk kirurg- kränkning av hållning. Deformitet i bröstet.
Genetiker Noonans syndrom.
Med hänsyn till barnets fenotyp, historikdata, resultaten av ytterligare studier, gjordes diagnosen Noonans syndrom, vilket bekräftades av resultatet av en molekylärgenetisk studie.
Således visar den presenterade kliniska observationen komplexiteten i differentialdiagnostisk sökning, behovet av att integrera individuella tecken i den allmänna fenotypen av ett visst patologiskt tillstånd för riktad snabb diagnos av vissa former av ärftliga sjukdomar och vikten av molekylärgenetiska metoder för att klargöra diagnos. Snabb diagnos, förtydligande av uppkomsten av varje syndrom är särskilt viktiga, eftersom de låter dig hitta den bästa metoden för behandling av dessa tillstånd, förebyggande av möjliga komplikationer (upp till barnets funktionsnedsättning); förebyggande av återfall av ärftliga sjukdomar i drabbade familjer (medicinsk genetisk rådgivning). Detta dikterar behovet av läkare av olika specialiteter att tydligt navigera i flödet av ärftlig patologi.
Bibliografi:
- Baird P., De Jong B. Noonans syndrom (XX och XY Turner-fenotyp) i tre generationer av en familj // J. Pediatr., 1972, vol. 80, sid. 110–114.
- Hasegawa T., Ogata T. et al. Koarktation av aorta och njurhupoplasi hos en pojke med Turner/Noonan ytanomalier och en 46, XY karyotyp: en klinisk modell för möjlig försämring av en förmodad lymfogen gen(er) för Turner somatisk stigmata // Hum. Genet., 1996, vol. 97, sid. 564–567.
- Fedotova T.V., Kadnikova V.A. et al. Klinisk-molekylär-genetisk analys av Noonans syndrom. Material från VI-kongressen av Russian Society of Medical Genetics. Medicinsk genetik, tillägg till nr 5, 2010, s.184.
- Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? // Br. J. Dermatol., 1994, vol. 131, sid. 270–274.
- Municchi G., Pasquino A.M. et al. Tillväxthormonbehandling vid Noonans syndrom: rapport om fyra fall som nådde slutlig höjd // Horm. Res., 1995, vol. 44, sid. 164–167.
Det finns olika medfödda patologier hos barn. Vissa av dem anses vara ganska vanliga. Men det finns också sällsynta sjukdomar. Deras förekomst är låg. Många har dock ganska allvarliga konsekvenser. En av dessa patologier är Noonans syndrom. Sedan, överväg det mer detaljerat. Artikeln kommer att beskriva hur och varför Noonans syndrom uppträder. Ett foto av sjukdomen kommer också att presenteras i texten.
Allmän information
Noonans syndrom är en ärftlig patologi. Den muterade PTPN11-genen från föräldrarna som bär den överförs till avkomman. Som regel är män infertila. Därför överförs genen genom moderlinjen. Den familjära karaktären av denna sjukdom noteras vanligtvis. Sällsynta fall som inte är relaterade till ärftlighet, men de registreras också i praktiken.
Historisk referens
En gång märkte Jacqueline Noonan, som praktiserade som barnkardiolog, medan hon arbetade på en klinik vid University of Iowa, att en viss grupp barn, som inkluderade både pojkar och flickor som hade lungstenos, ofta skilde sig åt i kortväxthet, storögda, simhudsförsedda halsar. , belägna låga öronbågar och ptos. Efter att ha undersökt kombinationen av manifestationer av hjärtsjukdom med andra utvecklingsavvikelser hos 833 patienter, skrev läkaren en artikel 1962. I sitt arbete beskrev hon nio fall där karakteristiska ansiktsdrag och kortväxthet noterades mot bakgrund av den medfödda typen. Sjukdomen hittades hos både män och kvinnor. 
Noonans syndrom: symtom
Det finns ett antal karakteristiska egenskaper som skiljer patologi från andra. Dessa inkluderar särskilt:
- Låg höjd. För kvinnor - 1,53, för män - 1,63 m. Vid födseln är både kroppslängden och barnets vikt inom det normala intervallet. Tillväxthämning börjar vid 2-3 års ålder.
- Ansiktsförändringar. Patienter som diagnostiserats med Noonans syndrom har vitt skilda former. Det finns ett hudveck i det inre hörnet. Det finns även ptos (hängande ögonlock) på grund av försvagad muskelfunktion eller på grund av små ögonhålor. Ungefär två tredjedelar av alla barn med patologi har skelning.När ptosen är begränsad och skelning utvecklas.
- Brott mot käkarnas struktur. Den övre är underutvecklad, det finns en välvd hög gom. I båda käkarna är tändernas läge felaktig.
- Låg landning eller deformation av öronen. Som ett resultat av denna anomali är hörseln nedsatt.
- Kort och vid hals.
- Sköldbröst med breda bröstvårtor.
- Deformitet i armbågsleden (medfödd).
- Platt fotad.
- Korta fingrar.
Missbildningar av inre organ
Noonans syndrom åtföljs oftast av störningar i det kardiovaskulära systemets aktivitet. Mot bakgrund av patologi upptäcks en förträngning (stenos) i lungstammen, liksom en defekt i interventrikulär septum. Den andra platsen är upptagen av anomalier i det genitourinära systemet. Så från njurarnas sida avslöjas defekter som hypoplasi (brist på vävnadsutveckling) eller frånvaron av en njure. När det gäller puberteten varierar den från normal till helt defekt. Flickor har ofta en sen menstruation, pojkar har kryptorkism eller testiklar är helt frånvarande. Det finns också kränkningar av spermatogenes. I vissa fall saknas spermier helt. Mot bakgrund av operationer med Noonans syndrom noteras ökad blödning. Vissa patienter kan också visa milda avvikelser. 
Blanketter
Det finns två typer av patologi:
- familjeform. Det skiljer sig i den ärftliga naturen hos den autosomalt dominanta typen. Hos bärare av mutantgenen uppträder avkomma med patologi.
- sporadisk form. I det här fallet uppträder mutationen från fall till fall. I det här fallet identifierades inte den ärftliga faktorn.
Orsaker
Den vanligaste faktorn som provocerar patologi är en mutation i PTPN11-genen. Denna orsak upptäcks hos 50 % av patienterna. Men hos en viss procent av personer med detta syndrom är den genetiska faktorn oklar. Arv av patologi förekommer i en autosomal dominant form. Syndromet kan utlösas av en ny mutation. Av detta följer att föräldrar med en sådan gen inte har någon större chans att få ett barn till.
Diagnos
Den är installerad i enlighet med de karakteristiska externa funktionerna (de beskrivs ovan). Laboratorieindikatorer undersöks också under diagnosen. I synnerhet finns det en minskning av koncentrationen av testosteron, koagulationsfaktor XII. Instrumentella studier genomförs också. En röntgen av bröstbenet, ekokardiografi, EKG föreskrivs. Med hjälp av dessa metoder upptäcks anomalier i inre organ. Läkaren kan också boka en konsultation 
Terapeutiska aktiviteter
På grund av syndromets genetiska tillstånd syftar behandlingen främst till att eliminera symtomen. Med kryptorkism föreskrivs en operation. Under det rör sig testiklarna in i pungen. Mot bakgrund av en minskning av koncentrationen av androgener rekommenderas hormonbehandling. I närvaro av njursvikt föreskrivs hemodialys. Med dess hjälp avlägsnas produkterna från metaboliska processer från kroppen genom extrarenal blodrening.
Noonans syndrom(J.A. Noonan, amerikansk endokrinolog, född 1928) är en ärftlig sjukdom som kännetecknas av kortväxthet och anomalier i somatisk utveckling. J. Nunen beskrivs mest fullständigt 1963. Den kan vara sporadisk eller familjär, nedärvd på ett autosomalt recessivt sätt. De patologiskt förändrade generna som orsakar N. av page är lokaliserade i autosomer (se. ärftliga sjukdomar ). Könskromatin och karyotyp Kromosomer ) matcha patientens kön. Sjukdomen förekommer hos både män och kvinnor.
De huvudsakliga kliniska manifestationerna av N. med. kortväxthet, nedre extremiteter, pterygoida hudveck på nacken, låg hårfäste på baksidan av huvudet, karakteristiska dermatoglyfiska förändringar, hypertelorism, ptos, antimongoloid snitt i ögonen, missbildning eller låg passform i öronkanterna, asymmetri i ansikte, en bred näsrygg, vilket gör patienter som liknar varandra till vän ( ris .). Karaktäriserad av en kort hals, medfödd deformitet i armbågsleden (cubitus valgus), brachydactyly, hög gom, patologisk bita , platta fötter, förändringar i kotornas form och struktur och andra anomalier noterade under Shereshevsky-Turners syndrom . Hittas ofta stenos i lungstammen, ventrikulär septumdefekt. Andra medfödda missbildningar i det kardiovaskulära systemet (se. Medfödda hjärtfel ) är mindre vanliga. Sexuell utveckling på N. sida. varierar från normal till fullständig gonadal dysgenes (se Hermafroditism ). Flickor har ofta en sen menstruation, pojkar - kryptorkism , hypogonadism . Histologisk undersökning av materialet som erhållits från testikelbiopsi avslöjar en minskning eller fullständig frånvaro av könsceller och Leydig-cellshyperplasi. Andra manifestationer av N. med. är gynekomasti och mental retardation. I blodserumet hos patienter, innehållet av testosteron, östrogener (se. könshormoner ) och gonadotropiner (se hypofyshormoner ) är normalt eller förändrats beroende på svårighetsgraden av gonadal dysgenes. Koncentrationen av tillväxthormon i blodet är normal. Karyotyp 46XX eller 46XV motsvarar gonad- och passkönet.
Diagnosen ställs på grundval av karakteristiska kliniska tecken och laboratoriedata. N. s. differentieras oftast med Shereshevsky-Turners syndrom ( tabell .).
Behandling i närvaro av tecken på hypogonadism består i ersättningsterapi med könshormonpreparat. Användningen av tillväxthormon är ineffektiv. Enligt indikationerna, kirurgisk korrigering av medfödda missbildningar, behandling av psykiska störningar i samband med mental retardation (se. Endokrina psykiska störningar ). Prognosen för livet är gynnsam. Särskild uppmärksamhet bör ägnas sysselsättning: valet av specialitet bör bidra till patienternas sociala anpassning, med hänsyn till deras mentala förmågor och fysiska funktionshinder.
Differentialdiagnostiska egenskaper hos Noonan och Shereshevsky-Turners syndrom
kliniska tecken |
Noonans syndrom |
Shereshevsky-Turners syndrom |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
sexuell utveckling |
Noonans syndrom kännetecknas av flera medfödda missbildningar som är typiska för patienter med Shereshevsky-Turners syndrom, med en normal karyotyp. Ibland identifieras syndromet med Bonnevie-Ulrichs syndrom, som har en liknande klinisk bild. Men med Bonnevie-Ulrichs syndrom finns det ingen hjärtsjukdom, vilket mycket ofta observeras vid Shereshevsky-Turners syndrom och Noonan. Hos alla patienter kan mer eller mindre uttalade symtom på hypogonadism upptäckas, även om vissa författare tror att med Noonans syndrom utvecklas könsorganen normalt hos vissa pojkar. Etiologi. Syndromet ärvs på ett autosomalt dominant sätt med låg genpenetrans. Fördelningen av syndromet är ganska stor (1:1000 - 1:5000), men det finns inget tydligt diagnostiskt kriterium i form av en onormal karyotyp, som hos patienter med Shereshevsky-Turners syndrom. I detta avseende upptäcks inte alla fall av sjukdomen. Hypotalamus-hypofys-gonadala relationer (patogenes) vid Noonans syndrom studeras praktiskt taget inte. Det finns bara en rapport om egenskaperna hos reproduktionssystemet hos en patient, på grundval av vilken det är svårt att göra generaliseringar. Vi undersökte innehållet av testosteron och gonadotropa hormoner i blodet, samt de funktionella reserverna i testiklarna hos 8 pojkar med Noonans syndrom. Testosteronnivåerna hos alla patienter var under det normala. Urinutsöndringen av 17-KS reducerades också. Dessa fynd pekar på testikelhormonbrist vid Noonans syndrom. Serumnivåer av testosteron och gonadotropa hormoner och urinutsöndring av 17KS (M±m) hos patienter med Noonans syndrom
Kombinationen av otillräcklig utsöndring av testosteron och höga nivåer av LH hos 4 patienter indikerar en primär lesion i testiklarna, huvudsakligen interstitialceller. Administrering av CG under 3 dagar vid en daglig dos på 1500 IE/m2 ledde dock till en signifikant ökning av testosteronnivåerna i blodet hos dessa pojkar. Kauschunsky et al. (1977) när de administrerade GnRH till en patient med Noonans syndrom, noterade de en uttalad ökning av utsöndringen av gonadotropiner, som hos patienter med primär hypogonadism, och samtidigt en positiv reaktion av testiklarna på CG. Det begränsade antalet observationer gör det svårt att tolka uppgifterna. Det kan antas att hos patienter är känsligheten hos Leydig-cellreceptorer för endogent gonadotropin reducerad, men testiklarnas reserver bevaras, vilket uttrycks i en ökning av testosteronsekretionen efter stimulering med höga doser av hCG. Hos patienter med låga testosteronnivåer och normala nivåer av gonadotropa hormoner i blodet är patogenesen av testikelinsufficiens troligen nära patogenesen för normogonadotrop hypogonadism. Theintz, Savagl (1982) beskrev 5 pojkar i åldrarna 11,8 - 16,5 år med Noonans syndrom. Alla hade tydliga tecken på hypogonadism och släpade efter i sexuell utveckling. I 4 av dem återgick den sexuella utvecklingen till det normala, och i två spontant och i två - efter behandling med testosteron. Endast hos en patient var behandling med CG och testosteron ineffektiv. Hos patienter med Noonans syndrom kan således hypogonadismens karaktär vara annorlunda, trots den gemensamma kliniska bilden. Otillräcklighet i reproduktionssystemet i dem kan vara resultatet av en primär defekt i gonaderna, dysfunktion i hypotalamus-hypofyssystemet och förändringar i testiklarnas känslighet för gonadotrop stimulering. Den kliniska bilden liknar kliniken för Shereshevsky-Turners syndrom. Här är ett utdrag ur en av patienterna vi observerade. Serezha L., 8 år, lades in på den pediatriska endokrinologiska avdelningen på sjukhuset. Rauhfus den 03/05/82 med eftersläpning i fysisk och mental utveckling. Han föddes från den andra graviditeten, som fortsatte med svår toxicos. Vid 7:e - 8:e graviditetsveckan drabbades mamman av en akut luftvägsvirusinfektion. Den första graviditeten slutade i ett missfall. Mamman lider av medfött Pojken föddes för tidigt (extraherades med kejsarsnitt). Kroppsvikt 3500 g, längd 51 cm.I neonatalperioden är tillståndet allvarligt. Vid 4 månaders ålder diagnostiserades en hjärtsjukdom; pojken släpade efter i fysisk och mental utveckling från första året. Frånvaron av testiklar i pungen upptäcktes vid födseln. Vid intagning, kroppslängd 124 cm (under medel), vikt 22 kg. Fysiken är felaktig - en bred kort hals, en asymmetrisk bröstkorg, en trattformad deformitet av bröstbenet; hög gom, hypertelorism, örondeformitet, fingerdefekter, brett åtskilda bröstvårtor. Penis utvecklas tillfredsställande, pungen är mycket liten, testiklarna är inte påtagliga. Intelligensen är måttligt reducerad. Karyotyp 46XY. Röntgen av skallen utan drag. Benåldern motsvarar 7 år. I blodserumet LH 4,33 mIU / ml, FSH 3,13 mIU / ml, testosteron 0,56 nmol / l (alla indikatorer är reducerade). Utsöndring 17KS 9,1 mmol/dag. Funktionstest med en enda injektion av hCG är negativt - blodtestosteron - 0,57 nmol/l, 17-KS - 8,87 mmol/dag; 3-dagars test är positivt - testosteron - 1,54 nmol / l, 17KS - 10,15 mmol / dag. Baserat på anamnesen, den kliniska presentationen och laboratoriedata ställdes en diagnos av Noonans syndrom. Ett positivt 3-dagarstest möjliggjorde behandling av CG. I avsaknad av effekt indikeras kirurgisk behandling av kryptorkism. "Störningar i sexuell utveckling hos pojkar", Utseende.
Blanketter
Orsaker
DiagnostikDiagnosen ställs på grundval av:
Behandling av Noonans syndrom
Komplikationer och konsekvenser
Förebyggande av Noonans syndrom
Liknande artiklar
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||