Cechy kinetyki reakcji enzymatycznej. Warunki reakcji enzymatycznej
Enzym E odwracalnie łączy się z substratem S, tworząc niestabilny, pośredni kompleks enzym-substrat ES, który pod koniec reakcji rozpada się, uwalniając enzym i produkty reakcji P.
Te pomysły stały się podstawą teoria „zamka na klucz” E. Fishera (1890). Struktura centrum aktywnego jest komplementarna do struktury molekularnej substratu, zapewniając tym samym wysoką specyficzność enzymu. W tworzeniu kompleksów enzym-substrat biorą udział wiązania wodorowe, oddziaływania elektrostatyczne i hydrofobowe, a w niektórych przypadkach także wiązania kowalencyjne i koordynacyjne.
D.Koshland został opracowany teoria „indukowanej korespondencji” (1958) . Zgodność przestrzenna pomiędzy strukturą substratu i centrum aktywnego enzymu powstaje w momencie ich wzajemnego oddziaływania, co można wyrazić wzorem „rękawica - ręka”. Substrat indukuje zmiany konformacyjne w cząsteczce enzymu w taki sposób, że centrum aktywne przyjmuje orientację przestrzenną niezbędną do związania substratu. Te. Enzym będzie występował tylko w aktywnej (rozciągniętej) formie T (rozciągliwej) w momencie przyłączania substratu, w przeciwieństwie do nieaktywnej formy R (relaksacyjnej).
Obecnie hipoteza Koshlanda jest stopniowo zastępowana hipoteza korespondencja topochemiczna. Zachowując podstawowe zasady teorii „indukowanej korespondencji”, wyjaśnia specyfikę działania enzymu poprzez rozpoznanie tej części substratu, która nie ulega zmianie podczas katalizy.
Podobnie jak inne katalizatory, enzymy z termodynamicznego punktu widzenia przyspieszają reakcje chemiczne poprzez zmniejszenie energii aktywacji.
Energia aktywacji to energia potrzebna do przekształcenia wszystkich cząsteczek mola substancji w stan aktywowany w danej temperaturze.
Zarówno reakcje katalizowane enzymatycznie, jak i niekatalizowane enzymami mają tę samą standardową zmianę energii swobodnej (ΔG). Jednakże reakcja enzymatyczna ma niższą energię aktywacji. Działając na szybkość reakcji, enzymy nie zmieniają położenia równowagi między reakcją do przodu i do tyłu, a jedynie przyspieszają jej początek.
2.3. Kinetyka reakcji enzymatycznych
Kinetyka enzymów bada wpływ charakteru chemicznego reagujących substancji (enzymów, substratów) oraz warunków ich oddziaływania (stężenie, pH, temperatura, obecność aktywatorów lub inhibitorów) na szybkość reakcji enzymatycznej. Szybkość reakcji enzymatycznej (V) mierzy się zmniejszeniem ilości substratu lub wzrostem produktu w jednostce czasu.
W katalizie enzymatycznej enzym (E) odwracalnie łączy się z substratem (S), tworząc niestabilny kompleks enzym-substrat (ES), który pod koniec reakcji rozpada się uwalniając enzym (E) i produkty reakcji (P) :
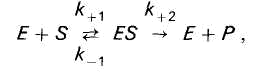
Ważną cechą reakcji enzymatycznych jest nasycenie enzymu substratem. Przy niskich stężeniach substratu szybkość reakcji jest wprost proporcjonalna do jego stężenia. Przy dużej prędkości szybkość reakcji jest maksymalna, staje się stała i niezależna od stężenia substratu [S] i jest całkowicie zdeterminowana przez stężenie enzymu (ryc. 11).
|
|
|
Ryż. 11. Zależność szybkości reakcji enzymatycznej od stężenia substratu przy stałym stężeniu enzymu. |

K S – stała dysocjacji kompleksu enzym-substrat ES, odwrotność stałej równowagi:
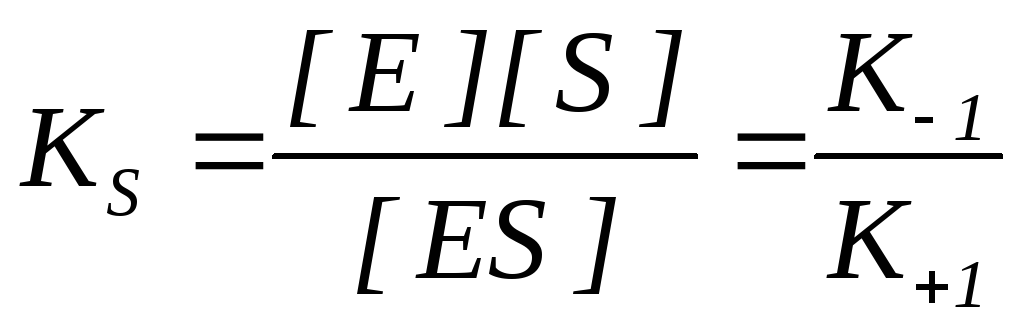
Im niższa wartość KS, tym większe powinowactwo enzymu do substratu.
Wyraża się ilościowa zależność między stężeniem substratu a szybkością reakcji enzymatycznej równanie Michaelisa-Menten:
 ,
,
– szybkość reakcji, Vmax – maksymalna prędkość reakcji enzymatycznej.
Briggs i Haldane poprawili równanie, wprowadzając Stała MichaelisaK M, ustalone eksperymentalnie.
Równanie Briggsa – Haldane’a: 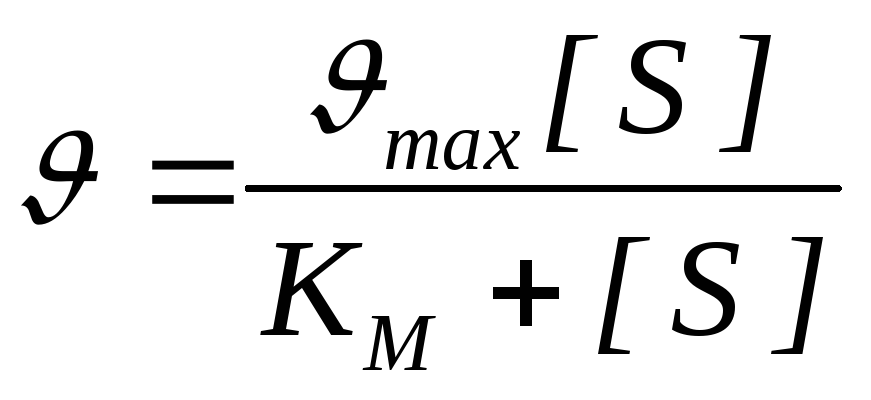
Stała Michaelisa jest liczbowo równa stężeniu substratu (mol/l), przy którym szybkość reakcji enzymatycznej jest o połowę mniejsza (ryc. 12). K m pokazuje powinowactwo enzymu do substratu; im niższa jego wartość, tym większe powinowactwo.
Eksperymentalne wartości K m dla większości reakcji enzymatycznych z udziałem jednego substratu wynoszą zwykle 10 -2 -10 -5 M. Jeśli reakcja jest odwracalna, wówczas oddziaływanie enzymu z substratem reakcji bezpośredniej charakteryzuje się K m różne od tego dla substratu reakcji odwrotnej.
OGÓLNA CHARAKTERYSTYKA ENZYMÓW
Enzymy są katalizatorami biologicznymi.
Charakter chemiczny enzymów. Miejsce aktywne enzymów.
Mechanizm katalizy enzymatycznej.
I. Enzymy – biologiczne katalizatory o charakterze białkowym, zdolne do wielokrotnego przyspieszania reakcje chemiczne napływające ciała, ale same nie są częścią końcowych produktów reakcji.
Nazywa się substancje, na które działa enzym podłoża.
Cała gama reakcji biochemicznych zachodzących w mikroorganizmach, roślinach i zwierzętach jest katalizowana przez odpowiednie enzymy. Wielka rola enzymów w technologii produkty żywieniowe. Produkcja każdego produktu spożywczego opiera się na procesach biochemicznych (enzymatycznych) lub fizycznych procesy chemiczne lub procesy te są ze sobą powiązane.
W przeciwieństwie do katalizatorów nieorganicznych, enzymy mają swoje własne cechy:
Szybkość katalizy enzymatycznej jest o kilka rzędów wielkości większa (od 10 3 do 10 9) niż w przypadku katalizatora niebiologicznego;
działanie każdego enzymu jest wysoce specyficzne, tj. każdy enzym działa tylko na swój własny substrat lub grupę pokrewnych substratów;
enzymy katalizują reakcje chemiczne łagodne warunki, tj. przy normalnym ciśnieniu, wysoka temperatura(20-50С) i przy pH środowiska, w większości przypadków zbliżonym do obojętnego.
Z punktu widzenia lokalizacji enzymów w komórce dzielimy je na zewnątrzkomórkowe i wewnątrzkomórkowe.
Zewnątrzkomórkowy enzymy są uwalniane przez żywą komórkę do środowiska zewnętrznego, wewnątrzkomórkowy – zlokalizowane są albo w organellach komórkowych, albo w kompleksach ze strukturami supramolekularnymi.
Specjalną grupę enzymów stanowią kompleksy wieloenzymowe, w skład których wchodzi szereg enzymów katalizujących kolejne reakcje transformacji dowolnego substratu. Kompleksy te są zlokalizowane w strukturach wewnątrzcząsteczkowych w taki sposób, że każdy enzym znajduje się w bliskiej odległości od enzymu katalizującego reakcję w łańcuchu danej sekwencji reakcji. Dzięki takiemu ułożeniu enzymów proces dyfuzji substratu i produktów reakcji jest zminimalizowany.
II. Enzymy są związkami białkowymi o dużej masie cząsteczkowej.
Podobnie jak inne białka, enzymy mają 4 poziomy budowy, mają wszystkie właściwości fizyczne i chemiczne białek, a tylko jedną charakterystyczną cechą jest zdolność do przyspieszania reakcji chemicznych. Enzymy mogą być proste - jednoskładnikowe i złożone dwuskładnikowe.
Enzymy jednoskładnikowe – zbudowane z łańcuchów polipeptydowych i podczas hydrolizy rozkładają się jedynie na aminokwasy.
Enzymy dwuskładnikowe – składają się z części białkowej – apoforma i część niebiałkowa - kofaktor. Obydwa składniki indywidualnie nie mają aktywności enzymatycznej. Tylko łącząc się ( holoenzym) uzyskują właściwości charakterystyczne dla biokatalizatorów. Rolę kofaktora może pełnić dowolny jon (Zn 2+, Mg 2+, Fe 2+, Cu 2+, rzadziej K + i Na +) lub związek organiczny(witaminy, nukleotydy). Organiczne kofaktory nazywane są koenzymy.
Rodzaj połączenia między kofaktorem a apoenzymem może być inny. W niektórych przypadkach występują one oddzielnie i wiążą się dopiero podczas reakcji; w innych przypadkach kofaktor i apoenzym są stale połączone, czasami silnymi wiązaniami kowalencyjnymi.
Miejsce aktywne enzymów– jest to lokalna część cząsteczki enzymu biorąca udział w akcie katalizy. W jednoskładnikowy W enzymach centrum aktywne powstaje w wyniku określonej orientacji reszt aminokwasowych łańcucha polipeptydowego. Zwykle w jego tworzeniu bierze udział niewielka liczba aminokwasów, w zakresie 12-16. Grupy funkcyjne tych aminokwasów mogą należeć do jednostek łańcucha polipeptydowego, które są od siebie odległe. Ich zbieżność związana jest z utworzeniem trzeciorzędowej struktury enzymu.
W dwuskładnikowy W enzymach centrum aktywne stanowi kompleks kofaktora i niektórych sąsiadujących reszt aminokwasowych.
W aktywnym ośrodku znajdują się kontakt(kotwica) obszar, którego funkcją jest wiązanie podłoża, oraz katalityczny – gdzie przemiana substratu w produkty reakcji następuje po związaniu go w miejscu kontaktu. W tworzeniu tych obszarów biorą udział: grupy funkcyjne: grupy COOH aminokwasów dikarboksylowych lub grupy końcowe łańcucha polipeptydowego; grupa imidazolowa histydyny; grupa OH seryny, NH 2 - grupa lizyny i grupy końcowe łańcucha polipeptydowego; grupa fenolowa tyrozyny i reszty hydrofobowe aminokwasów alifatycznych.
III. Określa się szybkość dowolnej reakcji enzymatycznej energiabariera, które reagujące cząsteczki muszą pokonać. Według Arrheniusa reakcję chemiczną z punktu widzenia energii procesu opisuje równanie
N = N 0 e -(E akt /RT) ,
gdzie N jest liczbą aktywnych cząsteczek, N 0 jest całkowitą liczbą reagujących cząsteczek; e – podstawa logarytmu naturalnego, R – stała gazowa, T – temperatura absolutna; Akt E – energia aktywacji.
Energia aktywacji- dodatkowa ilość energii potrzebna wszystkim cząsteczkom do pokonania bariery energetycznej reakcji i wejścia do niej. Energia ta jest różnicą pomiędzy całkowitą energią reagujących cząsteczek a energią wzbudzony stan przejściowy. Im wyższa energia aktywacji w reagującym układzie, tym wyższa bariera energetyczna i mniejsza szybkość reakcji.
Najważniejszą funkcją enzymu jest spadek energii aktywacji proces katalizowany. Na ryc. 1 przedstawia wykres zmian energii substancji nieenzymatycznej (1) i enzymatyczny (2) reakcje. Enzym zmniejsza wysokość bariery energetycznej (akt E E akt).
Mechanizm katalizy enzymatycznej pozostaje w dużej mierze niejasny. Jednakże zasadniczą rolę w powstaniu kinetyki enzymatycznej odegrały prace M. Michaelisa i M. Mentena, w których idea kompleks enzym-substrat. Powstawanie tego kompleksu prowadzi do spadku energii aktywacji reakcji.
Proces katalizy enzymatycznej można podzielić na trzy etapy:
Przestrzenne wiązanie substratu S z miejscem aktywnym enzymu E (tworzenie kompleksu enzym-substrat ES).
Konwersja kompleksu pierwotnego ES w aktywowany kompleks przejściowy ES ≠.
Oddział produkt finalny Reakcje P z enzymu.
Pierwszy etap jest krótki i zależny od stężenia substratu i enzymu w ośrodku, od szybkości dyfuzji substratu do centrum aktywnego enzymu. W tworzeniu kompleksu ES mogą brać udział zarówno wiązania kowalencyjne, koordynacyjne, jonowe, jak i słabsze formy wiązań w różnych kombinacjach - przyciąganie elektrostatyczne grup polarnych, siły kohezji van der Waalsa pomiędzy niepolarnymi sekcjami cząsteczek, wiązania wodorowe. O charakterze tych wiązań decydują właściwości chemiczne zarówno substratu, jak i grup funkcyjnych wchodzących w skład centrum aktywnego enzymu.
Drugi etap to tak naprawdę akt katalizy, tj. akt zerwania lub utworzenia nowych wiązań w podłożu; jest najwolniejszy i ogranicza szybkość reakcji chemicznej. Na tym etapie energia aktywacji reakcji enzymatycznej maleje z powodu tworzenie aktywnego kompleksu przejściowego ES ≠ .
NA Poziom molekularny pozwala lepiej zrozumieć mechanizm działania enzymów teoria katalizy kwasowo-zasadowej. Każda reakcja, która wiąże się z luką wiązania kowalencyjne, polega na udziale dwóch elementów elektronicznych o przeciwstawnym charakterze. Elektrony zrywanego wiązania muszą być przyciągane do składnika elektrofilowego, a z dala od składnika nukleofilowego. Odczynnikami, które mogą powodować takie przegrupowanie elektronów, są kwas i zasada. Nie jest jednak możliwe jednoczesne wytworzenie wysokich stężeń obu składników w tym samym roztworze, gdyż wzajemnie się neutralizują. W cząsteczce białka enzymu, dzięki konsolidacja W miejscu katalitycznym grup elektrofilowych i nukleofilowych nie zachodzi bezpośrednia reakcja zobojętniania. To w rzeczywistości determinuje akt katalizy. Znajdujące się w pewnej odległości od siebie grupy elektrofilowe i nukleofilowe miejsca katalitycznego enzymu nie tylko wiążą się z grupami reagującymi substratu, ale także wywierają silny wpływ polaryzujący na grupy substratu. Do tego należy dodać możliwość wahań ładunku w kompleksie ES, co stwarza wysoki stopień efektywności tej polaryzacji. Jest to przyczyną spadku energii aktywacji podczas katalizy enzymatycznej.
Zgodnie z teorią kowalencyjnykataliza niektóre enzymy oddziałują ze swoimi substratami, tworząc niestabilne, kowalencyjnie związane kompleksy enzym-substrat. Z tych kompleksów produkty reakcji powstają podczas kolejnej reakcji, znacznie szybciej niż w przypadku reakcji niekatalizowanych.
Zatem trzeci etap, kończący się utworzeniem produktów reakcji, zapewniają procesy zachodzące w poprzednich etapach.
OGÓLNA CHARAKTERYSTYKA ENZYMÓW
Enzymy są katalizatorami biologicznymi.
Charakter chemiczny enzymów. Miejsce aktywne enzymów.
Mechanizm katalizy enzymatycznej.
I. Enzymy – biologiczne katalizatory o charakterze białkowym, zdolne do wielokrotnego przyspieszania reakcji chemicznych zachodzących w ciała, ale same nie są częścią końcowych produktów reakcji.
Nazywa się substancje, na które działa enzym podłoża.
Cała gama reakcji biochemicznych zachodzących w mikroorganizmach, roślinach i zwierzętach jest katalizowana przez odpowiednie enzymy. Rola enzymów w technologii żywności jest ogromna. Produkcja dowolnego produktu spożywczego opiera się na procesach biochemicznych (enzymatycznych) lub fizykochemicznych lub procesy te są ze sobą powiązane.
W przeciwieństwie do katalizatorów nieorganicznych, enzymy mają swoje własne cechy:
Szybkość katalizy enzymatycznej jest o kilka rzędów wielkości większa (od 10 3 do 10 9) niż w przypadku katalizatora niebiologicznego;
działanie każdego enzymu jest wysoce specyficzne, tj. każdy enzym działa tylko na swój własny substrat lub grupę pokrewnych substratów;
enzymy katalizują reakcje chemiczne w łagodnych warunkach, tj. przy normalnym ciśnieniu, wysokiej temperaturze (20-50С) i przy pH środowiska, w większości przypadków zbliżonym do obojętnego.
Z punktu widzenia lokalizacji enzymów w komórce dzielimy je na zewnątrzkomórkowe i wewnątrzkomórkowe.
Zewnątrzkomórkowy enzymy są uwalniane przez żywą komórkę do środowiska zewnętrznego, wewnątrzkomórkowy – zlokalizowane są albo w organellach komórkowych, albo w kompleksach ze strukturami supramolekularnymi.
Specjalną grupę enzymów stanowią kompleksy wieloenzymowe, w skład których wchodzi szereg enzymów katalizujących kolejne reakcje transformacji dowolnego substratu. Kompleksy te są zlokalizowane w strukturach wewnątrzcząsteczkowych w taki sposób, że każdy enzym znajduje się w bliskiej odległości od enzymu katalizującego reakcję w łańcuchu danej sekwencji reakcji. Dzięki takiemu ułożeniu enzymów proces dyfuzji substratu i produktów reakcji jest zminimalizowany.
II. Enzymy są związkami białkowymi o dużej masie cząsteczkowej.
Podobnie jak inne białka, enzymy mają 4 poziomy budowy, mają wszystkie właściwości fizyczne i chemiczne białek, a tylko jedną charakterystyczną cechą jest zdolność do przyspieszania reakcji chemicznych. Enzymy mogą być proste - jednoskładnikowe i złożone dwuskładnikowe.
Enzymy jednoskładnikowe – zbudowane z łańcuchów polipeptydowych i podczas hydrolizy rozkładają się jedynie na aminokwasy.
Enzymy dwuskładnikowe – składają się z części białkowej – apoforma i część niebiałkowa - kofaktor. Obydwa składniki indywidualnie nie mają aktywności enzymatycznej. Tylko łącząc się ( holoenzym) uzyskują właściwości charakterystyczne dla biokatalizatorów. Rolę kofaktora może pełnić dowolny jon (Zn 2+, Mg 2+, Fe 2+, Cu 2+, rzadziej K + i Na +) lub związek organiczny (witaminy, nukleotydy). Organiczne kofaktory nazywane są koenzymy.
Rodzaj połączenia między kofaktorem a apoenzymem może być inny. W niektórych przypadkach występują one oddzielnie i wiążą się dopiero podczas reakcji; w innych przypadkach kofaktor i apoenzym są stale połączone, czasami silnymi wiązaniami kowalencyjnymi.
Miejsce aktywne enzymów– jest to lokalna część cząsteczki enzymu biorąca udział w akcie katalizy. W jednoskładnikowy W enzymach centrum aktywne powstaje w wyniku określonej orientacji reszt aminokwasowych łańcucha polipeptydowego. Zwykle w jego tworzeniu bierze udział niewielka liczba aminokwasów, w zakresie 12-16. Grupy funkcyjne tych aminokwasów mogą należeć do jednostek łańcucha polipeptydowego, które są od siebie odległe. Ich zbieżność związana jest z utworzeniem trzeciorzędowej struktury enzymu.
W dwuskładnikowy W enzymach centrum aktywne stanowi kompleks kofaktora i niektórych sąsiadujących reszt aminokwasowych.
W aktywnym ośrodku znajdują się kontakt(kotwica) obszar, którego funkcją jest wiązanie podłoża, oraz katalityczny – gdzie przemiana substratu w produkty reakcji następuje po związaniu go w miejscu kontaktu. W tworzeniu tych sekcji biorą udział następujące grupy funkcyjne: grupy COOH aminokwasów dikarboksylowych lub grupy końcowe łańcucha polipeptydowego; grupa imidazolowa histydyny; grupa OH seryny, NH 2 - grupa lizyny i grupy końcowe łańcucha polipeptydowego; grupa fenolowa tyrozyny i reszty hydrofobowe aminokwasów alifatycznych.
III. Określa się szybkość dowolnej reakcji enzymatycznej energiabariera, które reagujące cząsteczki muszą pokonać. Według Arrheniusa reakcję chemiczną z punktu widzenia energii procesu opisuje równanie
N = N 0 e -(E akt /RT) ,
gdzie N jest liczbą aktywnych cząsteczek, N 0 jest całkowitą liczbą reagujących cząsteczek; e – podstawa logarytmu naturalnego, R – stała gazowa, T – temperatura bezwzględna, Akt E – energia aktywacji.
Energia aktywacji- dodatkowa ilość energii potrzebna wszystkim cząsteczkom do pokonania bariery energetycznej reakcji i wejścia do niej. Energia ta jest różnicą pomiędzy całkowitą energią reagujących cząsteczek a energią wzbudzony stan przejściowy. Im wyższa energia aktywacji w reagującym układzie, tym wyższa bariera energetyczna i mniejsza szybkość reakcji.
Najważniejszą funkcją enzymu jest spadek energii aktywacji proces katalizowany. Na ryc. 1 przedstawia wykres zmian energii substancji nieenzymatycznej (1) i enzymatyczny (2) reakcje. Enzym zmniejsza wysokość bariery energetycznej (akt E E akt).
Mechanizm katalizy enzymatycznej pozostaje w dużej mierze niejasny. Jednakże zasadniczą rolę w powstaniu kinetyki enzymatycznej odegrały prace M. Michaelisa i M. Mentena, w których idea kompleks enzym-substrat. Powstawanie tego kompleksu prowadzi do spadku energii aktywacji reakcji.
Proces katalizy enzymatycznej można podzielić na trzy etapy:
Przestrzenne wiązanie substratu S z miejscem aktywnym enzymu E (tworzenie kompleksu enzym-substrat ES).
Konwersja kompleksu pierwotnego ES w aktywowany kompleks przejściowy ES ≠.
Oddzielenie końcowego produktu P reakcji od enzymu.
Pierwszy etap jest krótki i zależny od stężenia substratu i enzymu w ośrodku, od szybkości dyfuzji substratu do centrum aktywnego enzymu. W tworzeniu kompleksu ES mogą brać udział zarówno wiązania kowalencyjne, koordynacyjne, jonowe, jak i słabsze formy wiązań w różnych kombinacjach - przyciąganie elektrostatyczne grup polarnych, siły kohezji van der Waalsa pomiędzy niepolarnymi sekcjami cząsteczek, wiązania wodorowe. O charakterze tych wiązań decydują właściwości chemiczne zarówno substratu, jak i grup funkcyjnych wchodzących w skład centrum aktywnego enzymu.
Drugi etap to tak naprawdę akt katalizy, tj. akt zerwania lub utworzenia nowych wiązań w podłożu; jest najwolniejszy i ogranicza szybkość reakcji chemicznej. Na tym etapie energia aktywacji reakcji enzymatycznej maleje z powodu tworzenie aktywnego kompleksu przejściowego ES ≠ .
Na poziomie molekularnym daje lepsze zrozumienie mechanizmu działania enzymów teoria katalizy kwasowo-zasadowej. Każda reakcja polegająca na zerwaniu wiązań kowalencyjnych wiąże się z udziałem dwóch elementów elektronicznych o przeciwstawnym charakterze. Elektrony zrywanego wiązania muszą być przyciągane do składnika elektrofilowego, a z dala od składnika nukleofilowego. Odczynnikami, które mogą powodować takie przegrupowanie elektronów, są kwas i zasada. Nie jest jednak możliwe jednoczesne wytworzenie wysokich stężeń obu składników w tym samym roztworze, gdyż wzajemnie się neutralizują. W cząsteczce białka enzymu, dzięki konsolidacja W miejscu katalitycznym grup elektrofilowych i nukleofilowych nie zachodzi bezpośrednia reakcja zobojętniania. To w rzeczywistości determinuje akt katalizy. Znajdujące się w pewnej odległości od siebie grupy elektrofilowe i nukleofilowe miejsca katalitycznego enzymu nie tylko wiążą się z grupami reagującymi substratu, ale także wywierają silny wpływ polaryzujący na grupy substratu. Do tego należy dodać możliwość wahań ładunku w kompleksie ES, co stwarza wysoki stopień efektywności tej polaryzacji. Jest to przyczyną spadku energii aktywacji podczas katalizy enzymatycznej.
Zgodnie z teorią kowalencyjnykataliza niektóre enzymy oddziałują ze swoimi substratami, tworząc niestabilne, kowalencyjnie związane kompleksy enzym-substrat. Z tych kompleksów produkty reakcji powstają podczas kolejnej reakcji, znacznie szybciej niż w przypadku reakcji niekatalizowanych.
Zatem trzeci etap, kończący się utworzeniem produktów reakcji, zapewniają procesy zachodzące w poprzednich etapach.
Kinetyka enzymów bada wpływ różnych czynników (stężeń S i E, pH, temperatury, ciśnienia, inhibitorów i aktywatorów) na szybkość reakcji enzymatycznych. Głównym celem badania kinetyki reakcji enzymatycznych jest uzyskanie informacji pozwalających na głębsze zrozumienie mechanizmu działania enzymów.
Krzywa kinetyczna pozwala określić początkową szybkość reakcji V 0 .
Krzywa nasycenia podłoża.
Zależność szybkości reakcji od stężenia enzymu.
Zależność szybkości reakcji od temperatury.
Zależność szybkości reakcji od pH.
|
|
Optymalne pH dla działania większości enzymów mieści się w granicach wartości fizjologiczne 6,0-8,0. Pepsyna jest aktywna przy pH 1,5-2,0, co odpowiada kwasowości soku żołądkowego. Arginaza, enzym specyficzny dla wątroby, jest aktywny przy 10,0. Wpływ pH na szybkość reakcji enzymatycznej jest związany ze stanem i stopniem jonizacji grup jonogennych w cząsteczkach enzymu i substratu. Czynnik ten determinuje konformację białka, stan centrum aktywnego i substratu, powstawanie kompleksu enzym-substrat i sam proces katalizy. |

Matematyczny opis krzywej nasycenia podłoża, stała Michaelisa .
|
|
Równanie opisujące krzywą nasycenia podłoża zostało zaproponowane przez Michaelisa i Mentona i nosi ich nazwy (równanie Michaelisa-Mentena): V = (V MAKS *[ S])/(km+[ S]) , gdzie Km jest stałą Michaelisa. Łatwo to obliczyć, gdy V = V MAX /2 Km = [S], tj. Km to stężenie substratu, przy którym szybkość reakcji wynosi ½ V MAX. Aby uprościć wyznaczanie V MAX i Km, można ponownie obliczyć równanie Michaelisa-Mentena. 1/V = (Km+[S])/(V MAKS *[S]), 1/V = Km/(V MAKS *[S]) + 1/V MAKS , |
|
|
1/ V = km/ V MAKS *1/[ S] + 1/ V MAKS Równanie Lineweavera-Burka. Równanie opisujące wykres Lineweavera-Burka jest równaniem linii prostej (y = mx + c), gdzie 1/V MAX jest punktem przecięcia prostej na osi y; Km/V MAX - tangens prostej; przecięcie prostej z osią odciętych daje wartość 1/Km. Wykres Lineweavera-Burka pozwala wyznaczyć Km na podstawie stosunkowo niewielkiej liczby punktów. Wykres ten wykorzystuje się także przy ocenie działania inhibitorów, co zostanie omówione poniżej. Wartość Km jest bardzo zróżnicowana: od 10 -6 mol/l dla enzymów bardzo aktywnych do 10 -2 dla enzymów o niskiej aktywności. |
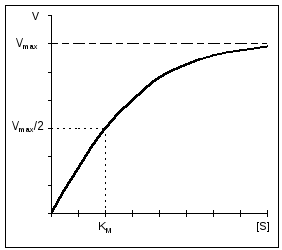
Szacunki km mają wartość praktyczną. Przy stężeniach substratu 100 razy większych niż Km, enzym będzie działał z prędkością bliską maksymalnej, więc maksymalna prędkość V MAX będzie odzwierciedlać ilość obecnego aktywnego enzymu. Okoliczność tę wykorzystuje się do oszacowania zawartości enzymu w preparacie. Ponadto Km jest cechą enzymu stosowaną do diagnozowania enzymopatii.
Hamowanie aktywności enzymów.
Niezwykle charakterystyczną i ważną cechą enzymów jest ich inaktywacja pod wpływem określonych inhibitorów.
Inhibitory - są to substancje powodujące częściowe lub całkowite zahamowanie reakcji katalizowanych przez enzymy.
Hamowanie aktywności enzymatycznej może być nieodwracalne lub odwracalne, konkurencyjne lub niekonkurencyjne.
Nieodwracalne zahamowanie - jest to trwała inaktywacja enzymu, wynikająca z kowalencyjnego związania cząsteczki inhibitora w miejscu aktywnym lub w innym specjalnym centrum, które zmienia konformację enzymu. Praktycznie wykluczona jest dysocjacja takich stabilnych kompleksów z regeneracją wolnego enzymu. Aby przezwyciężyć konsekwencje takiego hamowania, organizm musi syntetyzować nowe cząsteczki enzymów.
Odwracalne hamowanie – charakteryzuje się równowagowym kompleksowaniem inhibitora z enzymem dzięki wiązaniom niekowalencyjnym, w wyniku czego kompleksy te mają zdolność do dysocjacji z przywróceniem aktywności enzymu.
Podział inhibitorów na konkurencyjne i niekonkurencyjne opiera się na tym, czy są one osłabione ( hamowanie konkurencyjne ) lub nie osłabione ( hamowanie niekonkurencyjne ) ich działanie hamujące, gdy wzrasta stężenie substratu.
Inhibitory konkurencyjne - są to z reguły związki, których budowa jest zbliżona do struktury podłoża. Dzięki temu mogą wiązać się w tym samym miejscu aktywnym co substraty, zapobiegając interakcji enzymu z substratem już na etapie wiązania. Po związaniu inhibitor może zostać przekształcony w produkt lub pozostać w miejscu aktywnym do czasu wystąpienia dysocjacji.
Odwracalne hamowanie konkurencyjne można przedstawić w formie diagramu:
E↔ E-I → E + P 1
S (nieaktywny)
Stopień hamowania enzymu określa się na podstawie stosunku stężeń substratu i enzymu.
Klasycznym przykładem tego typu hamowania jest hamowanie aktywności dehydrogenazy bursztynianowej (SDH) przez jabłczan, który wypiera bursztynian z miejsca substratu i zapobiega jego konwersji do fumaranu:

Kowalencyjne wiązanie inhibitora z miejscem aktywnym powoduje inaktywację enzymu (nieodwracalne hamowanie). Przykład nieodwracalne hamowanie konkurencyjne może służyć do inaktywacji izomerazy triozofosforanowej za pomocą fosforanu 3-chloroacetolu. Inhibitor ten jest strukturalnym analogiem substratu, fosforanu dihydroksyacetonu, i nieodwracalnie wiąże się z resztą kwasu glutaminowego w miejscu aktywnym:
Niektóre inhibitory działają mniej selektywnie, oddziałując z określoną grupą funkcyjną w miejscu aktywnym różnych enzymów. Zatem wiązanie jodooctanu lub jego amidu z grupą SH aminokwasu cysteiny, zlokalizowaną w centrum aktywnym enzymu i biorącą udział w katalizie, prowadzi do całkowitej utraty aktywności enzymu:
R-SH + JCH 2 COOH → HJ + R-S-CH 2 COOH
Dlatego te inhibitory inaktywują wszystkie enzymy, które mają grupy SH zaangażowane w katalizę.
Nieodwracalne hamowanie hydrolaz pod wpływem gazów nerwowych (saryna, soman) wynika z ich kowalencyjnego wiązania z resztą seryny w centrum aktywnym.
Metoda hamowania konkurencyjnego znalazła szerokie zastosowanie w praktyka lekarska. Przykładem metabolizowanych inhibitorów kompetycyjnych mogą być leki sulfonamidowe, antagoniści kwasu p-aminobenzoesowego. Wiążą się z syntetazą dihydropterianową, enzymem bakteryjnym przekształcającym p-aminobenzoesan w kwas foliowy niezbędny do wzrostu bakterii. Bakteria ginie w wyniku przekształcenia związanego sulfanilamidu w inny związek kwas foliowy nie uformowany.
Inhibitory niekonkurencyjne zwykle wiążą się z cząsteczką enzymu w miejscu innym niż miejsce wiązania substratu, a substrat nie konkuruje bezpośrednio z inhibitorem. Ponieważ inhibitor i substrat wiążą się z różnymi centrami, możliwe jest utworzenie zarówno kompleksu E-I, jak i kompleksu SEI. Kompleks S-E-I również rozpada się, tworząc produkt, ale w wolniejszym tempie niż E-S, więc reakcja spowolni, ale się nie zatrzyma. Zatem mogą zachodzić następujące równoległe reakcje:
E↔ E-I ↔ S-E-I → E-I + P
Odwracalne hamowanie niekonkurencyjne jest stosunkowo rzadkie.
Inhibitory niekonkurencyjne nazywane są allosteryczny w odróżnieniu od konkurencyjnych ( izosteryczny ).
Odwracalne hamowanie można badać ilościowo za pomocą równania Michaelisa-Mentena.
Przy hamowaniu konkurencyjnym V MAX pozostaje stały, a Km wzrasta.
|
|
|
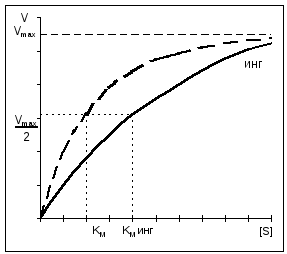
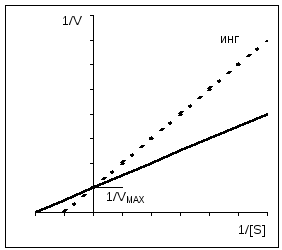
W przypadku hamowania niekonkurencyjnego V MAX zmniejsza się, podczas gdy Km pozostaje niezmieniona.
|
|
|

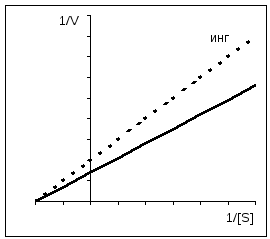
Jeśli produkt reakcji hamuje enzym, który katalizuje jego powstawanie, nazywa się tę metodę hamowania retroinhibicja Lub hamowanie sprzężenia zwrotnego . Na przykład glukoza hamuje glukozo-6-fosfatazę, która katalizuje hydrolizę glukozo-6-fosforanu.
Biologiczne znaczenie tego hamowania polega na regulacji niektórych szlaków metabolicznych (patrz następna lekcja).
CZĘŚĆ PRAKTYCZNA
Zadanie dla studentów
1. Badanie denaturacji białek pod wpływem roztworów kwasów mineralnych i organicznych oraz podczas ogrzewania.
2. Wykryj koenzym NAD w drożdżach.
3. Oznaczyć aktywność amylazy w moczu (surowicy krwi).
9. STANDARDY ODPOWIEDZI NA PROBLEMY, pytania testowe służące do kontroli wiedzy na zajęciach (można wykorzystać jako dodatek)
10. CHARAKTER I ZAKRES MOŻLIWYCH PRAC EDUKACYJNO-BADAWCZYCH NA TEMAT
(Wskazać konkretnie charakter i formę UIRS: przygotowywanie prezentacji abstrakcyjnych, prowadzenie samodzielnych badań, gry symulacyjne, sporządzanie wywiadu chorobowego z wykorzystaniem literatury monograficznej i innych form)
Wyślij swoją dobrą pracę do bazy wiedzy jest prosta. Skorzystaj z poniższego formularza
Studenci, doktoranci, młodzi naukowcy, którzy wykorzystują bazę wiedzy w swoich studiach i pracy, będą Państwu bardzo wdzięczni.
Opublikowano na http://www.allbest.ru/
ABSTRAKCYJNY
Enzymy i reakcje enzymatyczne
Enzymy
Aktywność enzymatyczna
Reakcja i specyficzność substratowa
Zajęcia enzymatyczne
Kataliza enzymatyczna
Kinetyka reakcji enzymatycznych
Inhibitory
Analiza enzymatyczna
enzym kataliza reakcji właściwości
Enzymy
Enzymy są biokatalizatory, tj. substancje pochodzenia biologicznego przyspieszające reakcje chemiczne. Uporządkowany ciąg procesów metabolicznych jest możliwy pod warunkiem zaopatrzenia każdej komórki w swój własny, genetycznie określony zestaw enzymów. Tylko pod tym warunkiem osiągana jest skoordynowana sekwencja reakcji. Enzymy biorą również udział w regulacji wielu z nich procesy metaboliczne, zapewniając w ten sposób, że metabolizm spełnia zmienione warunki. Prawie wszystkie enzymy są białka. Znane są również substancje aktywne katalitycznie kwasy nukleinowe– „rybozymy”.
Aktywność enzymatyczna
Katalityczne działanie enzymu, tj. jego działalność, określone w warunkach standardowych wg zwiększyć prędkość (fioletowy na schemacie) reakcja katalityczna ( kolor pomarańczowy) w porównaniu z niekatalitycznym ( żółty). Zwykle szybkość reakcji podaje się jako zmiana stężenia substratu lub produktu na jednostkę czasu(mol/(l s)). Ponieważ aktywność katalityczna nie zależy od objętości roztworu, w którym zachodzi reakcja, aktywność enzymatyczną wyraża się w katale; 1 kot to ilość enzymu, która przekształca 1 mol substratu w ciągu 1 sekundy. Inną jednostką aktywności jest jednostka międzynarodowa(E) - ilość enzymu, która przekształca 1 µmol substratu w ciągu 1 minuty (1 E = 16,7 ncat).
Reakcja i specyficzność substratowa
Akcja większości enzymy wysoki konkretnie. Pojęcie specyficzności nie dotyczy tylko typów reakcji katalitycznych ( specyficzność reakcji), ale także do natury związków - substratów ( specyficzność substratu). Jako przykład schemat pokazuje enzymy rozkładające wiązanie chemiczne. Wysoce specyficzne enzymy (typ A - górna linia tabeli) katalizują rozszczepienie tylko jednego rodzaju wiązania w substratach o określonej strukturze. Enzymy typu B (środkowy rząd) mają ograniczoną specyficzność reakcji, ale szeroką specyficzność substratową. Enzymy typu B (niska reakcja i niska specyficzność substratowa; podsumowanie) są rzadkie.
Zajęcia enzymatyczne
Do chwili obecnej znanych jest około 2000 różnych enzymów. Opracowany system klasyfikacji uwzględnia reakcję i specyficzność substratową enzymów. Wszystkie enzymy znajdują się w „Katalogu enzymów” pod ich numer klasyfikacyjny(KF), składający się z czterech cyfr. Pierwsza cyfra oznacza przynależność do jednego z sześć głównych klas. Kolejne dwie określają podklasę i podklasę, a ostatnia cyfra to numer enzymu w tej podklasie. Na przykład dehydrogenaza mleczanowa ma numer CF 1.1.1.27 (klasa 1, oksydoreduktazy; podklasa 1.1, dawca elektron - CH-OH; podklasa 1.1.1, akceptor--NADP+.)
Każda z sześciu głównych klas zawiera enzymy o tej samej specyficzności reakcji. OksydoreduktazaS(klasa 1) katalizują reakcje redoks. Transferazy(klasa 2) przenosi jedną lub drugą grupę funkcyjną z jednego podłoża na drugie. Oksydoreduktazy i transferazy wymagają wspólnego koenzymu. Hydrolazy(klasa 3) również uczestniczą w przeniesieniu grupowym, ale akceptorem jest tutaj zawsze cząsteczka wody. Liazy(klasa 4, czasami nazywane „syntezami”) katalizują rozszczepienie lub tworzenie związków chemicznych, w których powstają lub niszczą wiązania podwójne.
Izomerazy(klasa 5) przenosi grupy w cząsteczce bez zmian ogólna formuła podłoże. Ligazy(„syntezy”, klasa 6) katalizują reakcje addycji zależne od energii i dlatego ich działanie wiąże się z hydrolizą trifosforanu nukleozydu (najczęściej ATP).
Kataliza enzymatyczna
Enzymy - wysoce skuteczne katalizatory. Zwiększają szybkość katalizowanej reakcji 10-12 razy lub więcej. Aby zrozumieć mechanizm katalizy enzymatycznej, warto najpierw rozważyć występowanie reakcji niekatalitycznej.
Reakcja niekatalityczna (przy braku enzymu)
Jako przykład rozważ reakcję typu A + B > C + D. Substancje A i B w roztworze są otoczone otoczką cząsteczek wody (powłoka nawilżająca) i pod wpływem ruchu termicznego poruszają się losowo. Mogą ze sobą reagować tylko wtedy, gdy zderzą się w korzystnej orientacji, co jest mało prawdopodobne i rzadkie.
Do tworzenia produktów C + D złożony Powinny powstać A-B, powstałe w wyniku zderzenia cząsteczek stan przejściowy, co zwykle wymaga znacznych energia aktywacji E A. Ponieważ tylko niewielka część może otrzymać tę energię kompleksy A-B, osiągnięcie stanu przejściowego jest przypadkiem jeszcze rzadszym niż pojawienie się kompleksu. W roztworze większość energii aktywacji jest wydawana pokonywanie skorup hydratacyjnych pomiędzy A i B, zbliżając do siebie odczynniki I inne procesy chemiczne, w którym biorą udział te odczynniki. W rezultacie przy braku katalizatora powstawanie produktów zachodzi niezwykle rzadko, a szybkość reakcji v jest znikoma, nawet gdy reakcja jest dopuszczalna termodynamicznie, tj. DG< 0
Reakcja enzymatyczna
Enzymy specyficznie wiążą odczynniki (ich substraty). aktywny ośrodek. W tym przypadku podłoża są zorientowane w taki sposób, aby uzyskać optymalną pozycję do powstania stanu przejściowego (1-3). Zbliżenie I wymagana orientacja odczynniki znacznie zwiększają prawdopodobieństwo powstania kompleks produkcyjny A-B. Dodatkowo związanie substratu w miejscu aktywnym skutkuje usunięciem powłoki hydratacyjnej substratu, w wyniku czego usuwanie cząsteczek wody W centrum aktywnym enzymu podczas katalizy powstają zupełnie inne warunki niż w roztworze (3-5). Jeszcze jeden ważny czynnik wynika z interakcji pomiędzy resztami aminokwasowymi białka i substratu (4). Zatem stan przejściowy w przypadku reakcji enzymatycznej wymaga mniejszej energii aktywacji. Ponadto wiele enzymów przenosi określone grupy z lub na substrat podczas katalizy. Transfer protonów jest szczególnie powszechny. Ten enzymatyczny kataliza kwasowo-zasadowa znacznie skuteczniejsze niż wymiana protonów na kwasy i zasady w roztworze. Często grupy chemiczne są kowalencyjnie przyłączone do reszt enzymów. Zjawisko to nazywa się kataliza kowalencyjna.. Podstawy katalizy enzymatycznej
Chociaż obecnie trudno jest określić ilościowo udział poszczególnych efektów katalitycznych, uważa się, że jest to czynnik decydujący stabilizacja stanu przejściowego w miejscu aktywnym enzymu. W tym przypadku najistotniejszym punktem jest silne związanie nie tyle podłoża, co jego stanu przejściowego. Stanowisko to potwierdza niezwykle wysokie powinowactwo wielu enzymów do analogów stanu przejściowego, co można wytłumaczyć prostą analogią mechaniczną (na schemacie po prawej): jeśli chcemy wytoczyć z miejsca metalowe kulki (odczynniki) EA (stan podłoża) do energetycznie wyższego stanu przejściowego, a następnie w EP (stan produktu) należy ustawić magnes (katalizator) w taki sposób, aby siła przyciągania działała nie na EA (a), ale na przejście stan (b).
Kinetyka reakcji enzymatycznych
Kinetyka Określa się przede wszystkim reakcję enzymatyczną (tj. zależność szybkości reakcji od jej warunków). właściwości katalizatora, przez co jest znacznie bardziej złożona niż kinetyka reakcji niekatalitycznych
Model Michaelisa-Mentena
Prowadzi do tego pełna analiza matematyczna reakcji enzymatycznej złożone równania, nieodpowiednie dla praktyczne zastosowanie. Najwygodniejszym modelem okazał się model prosty, opracowany w 1913 roku. Wyjaśnia on charakterystyczną hiperboliczną zależność aktywności enzymu od stężenia substratu (1) i pozwala otrzymać stałe ilościowo charakteryzujące wydajność enzymu.
Model Michaelisa-Mentena wynika z tego, że początkowo substrat A tworzy kompleks z enzymem E (3), który znacznie szybciej ulega przekształceniu w produkt B niż w przypadku braku enzymu. Stała szybkości kcat (2) jest znacznie wyższa niż stała reakcji niekatalitycznej k. Stała k cat jest również nazywana „ Liczba rewolucji„ponieważ odpowiada liczbie cząsteczek substratu zamienionych w produkt przez jedną cząsteczkę enzymu w ciągu 1 s. Według tego modelu o aktywności enzymu decyduje proporcja kompleksu EA do całkowitego stężenia enzymu [E]t, czyli stosunek / [E]t (3). Dla uproszczenia model zakłada, że występują E, A i EA równowaga chemiczna zgodnie z prawem działania mas, które ostatecznie daje równanie dysocjacji kompleksu EA:
[E][A]/ = K m Ponieważ [E] t = [E] + ,
= [E] t [A]/(K m + [A])
Z v = k kat (2) i poprzedniego wyrażenia otrzymujemy Równanie Michaelisa-Mentena (4).
Równanie zawiera dwie wielkości ( dwa parametry), które nie zależą od stężenia substratu [A], ale charakteryzują właściwości enzymu: jest to produkt k cat [E] t, odpowiadający maksymalna prędkość reakcji V przy wysokich stężeniach substratu oraz Stała Michaelisa M, charakteryzujący podobieństwo enzym do substratu. Stała Michaelisa jest liczbowo równa stężeniu substratu [A].przy którym n osiąga połowę wartości maksymalnej V (jeśli v = V/2, to [A] / (K m + [A]) = 1/2, tj. K m = [A]). Charakteryzuje się wysokim powinowactwem enzymu do substratu niska wartość do m i odwrotnie,
Model Michaelisa-Mentena opiera się na kilku nie do końca realistycznych założeniach, takich jak nieodwracalna konwersja EA do E + B, osiągnięcie równowagi pomiędzy E, A i EA, brak innych form enzymu w roztworze poza E i EA. Tylko z zastrzeżeniem tych hipotetycznych warunków K m odpowiada stałej dysocjacji kompleksu, a k cat odpowiada stałej szybkości reakcji EA > E + B.
Definicja V i K M
Zasadniczo V i K m można wyznaczyć z wykresu v w funkcji [A] (ryc. po lewej). Od w asymptotycznie osiąga V wraz ze wzrostem stężenia substratu [A], trudno jest uzyskać wiarygodną wartość V i K m (ryc. po lewej) w drodze ekstrapolacji.
Dla wygody obliczeń równanie Michaelisa-Mentena można przekształcić tak, aby punkty eksperymentalne leżały na linii prostej. Dzięki jednej z takich przekształceń graficznych w tzw Wykres Eadie-Hofstee(zdjęcie po prawej) wykreśl zależność v od v/[A]. W tym przypadku punkt przecięcia prostej uzyskanej przez najlepsze liniowe przybliżenie punktów doświadczalnych z osią rzędnych odpowiada V, a tangens kąta nachylenia jest równy -K m. To graficzne podejście do wyznaczania V i K m również nie jest optymalne. Obecnie dane dotyczące kinetyki enzymów są przetwarzane szybciej i bardziej obiektywnie przy użyciu technologii komputerowej.
Inhibitory
Wiele związków może wpływać na metabolizm poprzez modulację aktywności odpowiednich enzymów. Zwłaszcza ważne funkcje jednocześnie spełnić inhibitory enzymów. Wiele inhibitorów enzymów tak substancje lecznicze pochodzenia naturalnego lub syntetycznego. Metabolity mogą być również inhibitorami enzymów w procesach regulacyjnych.
Rodzaje hamowania
Większość inhibitorów enzymów działa odwracalny, czyli nie wprowadzają żadnych zmian w cząsteczce enzymu po ich dysocjacji. Jednak są też nieodwracalny inhibitory enzymów, które nieodwracalnie modyfikują enzym docelowy. Zasada działania inhibitora rodzaj hamowania określić poprzez porównanie kinetyki reakcji w obecności inhibitorów bez niej (patrz diagram B). Wyróżnić konkurencyjny(A, po lewej) i niekonkurencyjny(Ah, tak) zahamowanie. Odgrywa ważną rolę w regulacji metabolizmu hamowanie allosteryczne(A, 6).
Tak zwana analogi substratów(2) mają właściwości podobne do właściwości docelowego substratu enzymu. Odwracalnie blokują część cząsteczek dostępnego enzymu, ale nie mogą być dalej przekształcane w produkt. Dlatego, aby osiągnąć połowę maksymalnej szybkości reakcji, więcej wysoki stężenie substratu: w obecności takiego inhibitora wzrasta stała Michaelisa K m (B). Podłoże w wysokie stężenia wypiera inhibitor z enzymu. Dlatego maksymalna prędkość V nie zmienia się przy tego rodzaju hamowaniu. Ponieważ substrat i inhibitor konkurują o miejsce wiązania enzymu, ten typ hamowanie nazywa się konkurencyjny. Analogi stanu przejściowego(3) działają również jako inhibitory konkurencyjne.
Jeśli inhibitor reaguje funkcjonalnie ważna grupa enzymu bez zakłócania wiązania substratu, takie hamowanie nazywa się niekonkurencyjny(na schemacie po prawej). W tym przypadku K m pozostaje niezmieniona, wręcz przeciwnie, zmniejsza się stężenie funkcjonalnie aktywnego enzymu [E] t, a w konsekwencji maksymalna szybkość reakcji V. Z reguły działają inhibitory niekonkurencyjne nieodwracalnie, ponieważ modyfikują grupy funkcyjne docelowego enzymu (4).
W przypadku tzw. „ substraty samobójcze" (5) mówimy o o analogach substratów zawierających dodatkową grupę reakcyjną. Początkowo wiążą się odwracalnie, a następnie tworzą związek kowalencyjny z miejscem aktywnym enzymu. Dlatego hamowanie przez takie związki objawia się jako niekonkurencyjny. Dobrze znanym przykładem takiego inhibitora jest antybiotyk penicylina.
Inhibitory allosteryczne wiążą się z poszczególnymi regionami enzymu poza centrum aktywnym (6). To połączenie pociąga za sobą zmiany konformacyjne w cząsteczce enzymu, co prowadzi do zmniejszenia jego aktywności. Efekty allosteryczne występują prawie wyłącznie w przypadku enzymy oligomeryczne. Kinetyki takich układów nie da się opisać prostym modelem Michaelisa-Mentena.
Kinetyka hamowania
Hamowanie konkurencyjne można łatwo odróżnić od hamowania niekonkurencyjnego, gdy jest stosowane Grafika Eady-Hofstee. Jak już wspomniano, konkurencyjny inhibitory wpływają tylko na Km, ale nie na V. Linie proste uzyskane na wykresie przy braku i obecności inhibitora przecinają się na osi rzędnych. Bezpośrednio dla niekonkurencyjny zahamowania mają to samo nachylenie (K m nie zmienia się), jednakże wraz ze wzrostem stężenia inhibitora odcinki odcięte tymi prostymi na osi rzędnych stają się coraz krótsze. W przypadku enzymów allosterycznych nie można zastosować wykresu Eady-Hofstee, który w tym przypadku jest nieliniowy (nie pokazano tutaj).
Analiza enzymatyczna
Enzymy odgrywają ważną rolę w analiza biochemiczna . W materiałach biologicznych, na przykład w płynach ustrojowych, enzymy można wykryć w znikomych stężeniach, oznaczając aktywność katalityczną. Enzymy można stosować jako odczynniki w celu określenia stężeń metabolitów, takich jak poziom glukozy we krwi (Rysunek B). Większość testów enzymatycznych wykorzystuje fotometrię.
Podstawy spektrofotometrii
Wiele cząsteczek absorbowaćświatło w widzialnym lub ultrafioletowym obszarze widma. Właściwość tę można wykorzystać do określenia stężeń. Stopień absorpcji zależy od rodzaju i stężenia substancji, a także długości fali użytego światła. Dlatego używają światło monochromatyczne, czyli światło o określonej długości fali, które można oddzielić od światła białego za pomocą monochromator. Światło monochromatyczne o natężeniu I 0 przechodzi przez prostokątną kuwetę wykonaną ze szkła lub kwarcu (kuweta), w której znajduje się roztwór substancji absorbującej. Natężenie I powstającego światła, osłabione przez absorpcję, mierzy się za pomocą detektora. Absorpcja światła(Rozwiązanie ( gęstość optyczna) definiuje się jako logarytm ujemny stosunku I / I 0 . Prawo Lamberta-Beera stwierdza, że A jest proporcjonalne do stężenia (c) substancji i grubości (d) warstwy roztworu. Współczynnik ekstynkcjimi zależy, jak wspomniano powyżej, od rodzaju substancji i długości fali.
Oznaczanie aktywności dehydrogenazy mleczanowej
Oznaczenie aktywności dehydrogenazy mleczanowej [LDH] opiera się na fakcie, że zredukowany koenzym NADH+H+ absorbuje światło o długości fali 340 nm, natomiast NAD+ nie wykazuje absorpcji przy tej długości fali. Widma absorpcyjne(tj. wykresy A w funkcji długości fali) substratu i koenzymu w reakcji LDH pokazano na ryc. B1.
Różnice w absorpcji NAD+ i NADH pomiędzy 300 a 400 nm wynikają ze zmian w pierścieniu nikotynamidowym podczas utleniania lub redukcji.
W celu określenia aktywności najpierw do kuwety umieszcza się roztwory mleczanu i NAD+ i rejestruje absorpcję o stałej długości fale 340 nm. Reakcja niekatalityczna przebiega z bardzo małą szybkością. Zatem mierzalne ilości NADH powstają dopiero po dodaniu LDH. Ponieważ szybkość wzrostu absorpcji DA/Dt zgodnie z prawem Lamberta-Beera jest proporcjonalna do szybkości reakcji DA/Dt, aktywność LDH można obliczyć za pomocą współczynnika ekstynkcji e przy 340 nm lub przez porównanie z roztworem wzorcowym.
Enzymatyczne oznaczanie glukozy
Większość biomolekuł nie absorbuje światła w zakresie widzialnym lub ultrafioletowym widma. Ponadto występują zwykle w mieszaninach z innymi związkami, które również powodują podobne reakcje chemiczne.
Obie trudności można pokonać odpowiedni enzym do selektywnej przemiany wyznaczonego metabolitu w substancję barwną, o czym dodatkowo decyduje intensywność absorpcji światła.
Typowa metoda oznaczania poziomu glukozy we krwi opiera się na dwóch kolejnych reakcjach:
1) tworzenie glukonolaktonu i nadtlenku wodoru H 2 O 2 pod działaniem enzymu oksydaza glukozowa ;
2) utlenianie bezbarwnej substancji pod wpływem nadtlenku wodoru do zabarwionego na zielono związku w reakcji katalizowanej przez peroksydazę.
Po zużyciu całej glukozy obecnej w próbce ilość utworzonej barwnej substancji można określić na podstawie absorpcji światła, która jest wprost proporcjonalna do początkowej zawartości glukozy.
Opublikowano na Allbest.ru
Podobne dokumenty
Przyspieszanie reakcji chemicznych za pomocą katalizatorów. Cechy enzymów (enzymów) jako wysoce specyficznych białek pełniących funkcje katalizatorów biologicznych. Budowa enzymów, ich specyfika i klasyfikacja. Etapy katalizy enzymatycznej.
prezentacja, dodano 20.11.2014
Klasyfikacja enzymów, ich funkcje. Konwencje nazewnictwa enzymów, budowa i mechanizm działania. Opis kinetyki reakcji enzymatycznych jednosubstratowych. Modele kluczowo-zamkowe korespondencji indukowanej. Modyfikacje, kofaktory enzymów.
prezentacja, dodano 17.10.2012
Charakterystyka enzymów, katalizatorów organicznych o charakterze białkowym przyspieszających reakcje niezbędne do funkcjonowania organizmów żywych. Warunki działania, produkcja i zastosowanie enzymów. Choroby związane z upośledzoną produkcją enzymów.
prezentacja, dodano 19.10.2013
Ogólna charakterystyka i główne typy enzymów. Właściwości chemiczne enzymy i reakcje, które katalizują. Selektywność i efektywność enzymów. Zależność od temperatury i środowiska roztworu. Miejsce aktywne enzymu. Szybkość reakcji enzymatycznych.
prezentacja, dodano 10.06.2014
Definicja enzymów jako specyficznych białek występujących we wszystkich żywych komórkach katalizatorów biologicznych. Przestrzenność cząsteczki strukturalnej enzymów, proces biosyntezy oksydoreduktazy, transferazy, hydrolazy, liazy, izomerazy i ligazy.
test, dodano 27.01.2011
Badania kinetyczne reakcji enzymatycznych w celu identyfikacji enzymów i porównania ich szybkości. Tworzenie kompleksu enzym-substrat z enzymu i substratu pod wpływem sił charakter fizyczny. Organizmy fakultatywne, autotrofy i heterotrofy.
test, dodano 26.07.2009
Enzymy: budowa biochemiczna i rola fizjologiczna. Analiza metod oznaczania aktywności enzymatycznej i widma enzymatycznego w płynach ustrojowych. Główne enzymy w moczu są normalne i patologiczne. Spektrum enzymów moczu w chorobach nerek.
raport, dodano 03.10.2015
Badanie enzymów, ich właściwości i mechanizmu działanie biologiczne. Przeprowadzać badanie nowoczesne pomysły o mechanizmie transaminacji enzymatycznej. Rozwój ogólna teoria kataliza pirydoksalna. Struktura kompleksu enzym-substrat.
streszczenie, dodano 14.03.2015
Specyficzne białka katalizujące reakcje chemiczne w układach żywych. Charakterystyka i klasyfikacja enzymów, ich wielkość i budowa. Wpływ warunków środowiskowych na aktywność enzymów: czynniki i kofaktory; choroby związane z zakłóceniem ich produkcji.
prezentacja, dodano 07.05.2015
Badanie przeznaczenia enzymów lub enzymów - cząsteczek białek lub cząsteczek RNA (rybozymów) lub ich kompleksów, które przyspieszają (katalizują) reakcje chemiczne w układach żywych. Lokalizacja enzymów w komórce. Dziedziczne i nabyte enzymopatie.
Podobne artykuły
-
Ciekawe fakty z życia Louisa de Funesa
Wielki francuski komik Louis de Funes nie miał nic wspólnego z wizerunkiem zabawnego głupca, który rozsławił go na ekranie. W życiu dziwactwa aktora nie przyniosły radości otaczającym go osobom. Cechy zrzędy, nudziarza i mizantropa można wyśledzić i...
-
Yuri Dud: biografia i życie osobiste dziennikarza
Do swojej pracy podchodzi odpowiedzialnie, jest to połączenie kanonicznego podejścia dziennikarskiego i wolnej osoby twórczej, co w skrócie można ująć w następujący sposób: „nieważne z kim wywiad, byle był ciekawy”. Yuri uważa test za udany...
-
Dziewczyna chwały dyskoteki Komunistycznej Partii Związku Radzieckiego
Prawdziwe imię i nazwisko: Alexandra Fedorov Rok urodzenia: 1993 Miejsce urodzenia: St. Petersburg Sasha Disco jest byłą dziewczyną rapera. Prawdziwe imię Sashy Discoteki to Fedorov. Sasza urodziła się w 1993 roku. Zainteresowanie osobowością Alexandry Discotheka...
-
Yaroslav Sumishevsky – przedstawiciel nowej generacji profesjonalnego wokalu
Z roku na rok zwiększa się grono wielbicieli talentu tego performera. Yaroslav Sumishevsky to muzyk i piosenkarz, którego popularność rośnie z każdym miesiącem, szczególnie w tym roku, kiedy on i jego grupa „Makhor-band” aktywnie...
-
Stanislav Belkovsky: biografia, działalność, rodzina i ciekawe fakty Belkovsky rozwiódł się z Olesją
Olesya Yakhno, którego biografia jest dość specyficzna, jest częstym gościem wielu rosyjskich talk show o tematyce politycznej. Pozycjonuje się jako ukraińska niezależna dziennikarka, ale nie jest to do końca prawdą. Olesya - lub Alesya - często...
-
Banosh, polenta, hominy i inne pyszne dania z mąki kukurydzianej
Kasza kukurydziana jest jedną z najbardziej przydatnych. Zawiera karoten (prowitaminę A), niezbędne dla naszego zdrowia witaminy B1, B2, C, PP, a także aminokwasy lizynę i tryptofan. Kasza kukurydziana nie tylko odżywia organizm, ale także...