Zespół Fanconiego u dzieci i dorosłych. Zespół Fanconiego: objawy, diagnostyka i metody leczenia Metody leczenia tego problemu
Zespół Fanconiego (pełna nazwa - de Toni-Debreu-Fanconi) jest wrodzoną patologią, która wyraża się w poważnej dysfunkcji proksymalnych kanalików nerkowych, a mianowicie naruszeniu wtórnego wchłaniania (wchłaniania do krwi) substancji filtrowanych przez nerki, co prowadzi do cukromoczu (zwiększonego poziomu cukru w moczu), fosfaturii (upośledzony metabolizm fosforu i wapnia), aminoacydurii (zwiększone wydalanie aminokwasów z moczem) i zmniejszenia stężenia wodorowęglanów regulujących kwasowość krwi.
Zespół De-Toniego-Debreu-Fanconiego
Zespół Fanconiego jest chorobą bardzo rzadką, występującą głównie u dzieci i według statystyk medycznych częstość jej występowania wynosi 1 chore dziecko na 350 tys. noworodków obu płci.
Obserwuje się go niezwykle rzadko u dorosłych, rozwijając się na tle nabytych patologii. Kod patologii według ICD-10: E72.O.
Powoduje
Charakter i przyczyny wady genetycznej zespołu Fanconiego nie są dziś dobrze poznane.
Przyjmuje się, że przyczyną patologii są albo defekty w białkach transportowych kanalików nerkowych, albo mutacja genu zaburzająca działanie enzymów regulujących wchłanianie zwrotne glukozy, aminokwasów i fosforu.
Istnieją dane badawcze dotyczące defektów punktowych w mitochondriach prowadzących do nieprawidłowego funkcjonowania kanalików nerkowych.
Choroba wiąże się także z nietolerancją fruktozy, przewlekłym zatruciem toksynami (metale ciężkie, ifosfamid, aminoglikozydy), niedoborem witaminy D, amyloidozą, niedoborem szeregu enzymów komórkowych (karboksylaza pirogronianowa, karboksykinaza fosfoenolopirogronianowa i inne), tyrozynemią, leukodystrofią metachromatyczną, galaktozemia, cystynoza, glikogenoza ami.
Według innych ekspertów zespół Fanconiego może być izolowaną patologią - a mianowicie jedną z ciężkich postaci patologii przypominających krzywicę, które mają charakter dziedziczny.
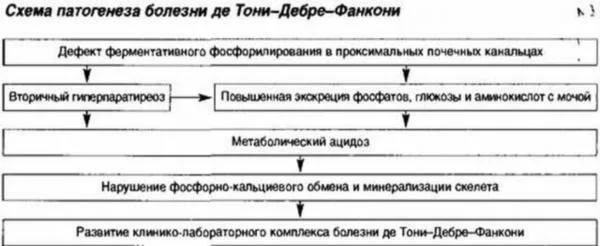
Badania potwierdzają, że w zespole Fanconiego zaburzony jest komórkowy metabolizm energetyczny obejmujący ATP (trifosforan adenozyny) oraz transport międzykomórkowy w kanalikach głównego elementu nerki – nefronu.
Na skutek niedostatecznej funkcji enzymów dochodzi do strat glukozy, fosforanów i aminokwasów, a w kanalikach nerkowych występuje niedobór energii. W tym przypadku z moczem wydalane są ważne substancje, co prowadzi do zmian zwyrodnieniowych tkanki kostnej – krzywicy.
Ponieważ medycyna nie doszła jeszcze do jednoznacznego wniosku co do przyczyn zespołu Faconiego, schorzenie to określa się także innymi terminami: „cukrzyca glukofosfaminowa”, „idiopatyczny nerkowy zespół Fanconiego”, „krzywica D-oporna”, „karłowatość nerkowa z D. krzywica oporna”, „dziedziczny zespół Fanconiego”.
Formy i patogeneza
Istnieją dwa rodzaje zespołu Fanconiego:
- dziedziczny (wrodzony, idiopatyczny), który odnosi się do formy pierwotnej;
- nabytą, co uważa się za wtórną postać choroby.
Eksperci kojarzą typ dziedziczny (genetyczny) z defektem chromosomu X, który dziedziczy się w sposób dominujący i recesywny, dlatego genetyczne przewidywanie jego ujawnienia się u przyszłego potomstwa nie jest zadaniem łatwym. Jeśli patologia jest typu wrodzonego (postać pierwotna), wówczas zostaje wykryta u dziecka w okresie niemowlęcym do jednego roku. Dlatego pierwotna forma nazywana jest „niemowlętem”.
Stopień mutacji genu determinuje również nasilenie samego zespołu. Zatem dziedziczny kompletny zespół Fanconiego objawia się obecnością 3 podstawowych defektów biochemicznych, do których należą cukromocz, aminoacyduria, fosfaturia, niepełny - w dwóch z nich.
Patologii uwarunkowanej genetycznie towarzyszą zazwyczaj inne schorzenia wrodzone: cystynoza, zespół Wilsona, zespół Denta, zespół Lowe’a, nietolerancja fruktozy, tyrozynemia (niezdolność organizmu do skutecznego rozkładania aminokwasu tyrozyny), galaktozemia (zaburzenie przemiany cukru w glukozę). ) i nadmierne gromadzenie glikogenu.
Rozwój zespołu Fanconiego
Zespół nabyty (wtórny), w przeciwieństwie do wrodzonego, nie towarzyszy, ale jest konsekwencją istniejących patologii:
- tyrozynemia typu I;
- cystynoza (zaburzony metabolizm aminokwasu cystyny z późniejszym uszkodzeniem nerek);
- nietolerancja fruktozy;
- choroba Wilsona-Kovalova;
- galaktozemia;
- glikogenoza (nieprawidłowe gromadzenie się glikogenu w tkankach i narządach) typ XI;
- dziedziczne patologie nerek;
- (zaburzenie metabolizmu białek prowadzące do stwardnienia rozsianego, atrofii, dysfunkcji narządów);
- cewkowo-śródmiąższowe (zapalenie nerek z uszkodzeniem tkanki, kanaliki nerkowe);
- nadczynność przytarczyc (choroba endokrynologiczna, w której zaburzony jest prawidłowy poziom wapnia i fosforu we krwi);
- nowotwory złośliwe: szpiczak, płuca, trzustka, płuca, choroby łańcuchów lekkich;
- głębokie oparzenia.
Ponadto zespół może być wywołany następującymi stanami:
- przeszczepianie narządów o niskiej zgodności tkankowej;
- niedobór witaminy D;
- zatrucie uranem, bizmutem, rtęcią, ołowiem, kadmem;
- kontakt z toluenem, kwasem maleinowym, lizolem;
- stosowanie nefrotoksycznych środków farmakologicznych, takich jak: gentamycyna, leki platynowe, przeterminowane leki na bazie tetracyklin, dydanozyna, cydofowir, przeciwnowotworowe leki chemioterapeutyczne – ifosfamid, streptozocyna.
Objawy i oznaki
W formie dziedzicznej (wrodzonej).
Objawy pierwotne pojawiają się w pierwszych miesiącach życia, rzadko po półtora roku.
Przede wszystkim u noworodka obserwuje się następujące stany:
- częste oddawanie moczu (wielomocz);
- zwiększone pragnienie (polidypsja);
- długotrwałe zaparcia;
- częste ataki bezprzyczynowych wymiotów;
- astenia (ogólne zmęczenie), osłabienie mięśni;
- niewyjaśnione „skoki” temperatury do 37,5 – 38 C;
- wzdęty brzuch.
Z reguły w okresie, gdy rozpoczynają się ataki wymiotów i wzrasta temperatura dziecka, dziecko zabierane jest do pediatry. Doświadczony specjalista musi ustalić, czy kombinacja objawów niepokojących rodziców nie ma związku z ostrymi infekcjami dróg oddechowych, ostrymi infekcjami wirusowymi dróg oddechowych lub infekcją enterowirusową.
Kompetentny pediatra jest w stanie w odpowiednim czasie rozpoznać zespół Fanconiego. Badania laboratoryjne mogą potwierdzić podejrzenia, jeśli ujawnią się trzy (lub dwa) podstawowe objawy: cukromocz, uogólniona hiperaminoacyduria i hiperfosfaturia, charakterystyczne dla tej patologii.
Po łagodnych i raczej niejasnych objawach w ciągu następnego półtora roku wyraźnie ujawniają się objawy charakterystyczne dla zespołu Fanconiego:
- Wczesny karłowatość (niskorosłość), spowodowana ciągłym usuwaniem z organizmu najważniejszych aminokwasów, glukozy, wapnia i fosforanów. Pierwsze sześć miesięcy normalnego wzrostu i masy ciała zostaje zastąpione deficytem masy ciała (do 30%) i wzrostu (od 2 do 21%).
- Krzywica, spowodowana masywnym wydalaniem wapnia i fosforanów, ujawnia się po 10–12 miesiącach życia i ma cechy charakterystyczne dla zespołu Fanconiego: główka dziecka jest zwykle lekko zdeformowana, ale duże kości nóg i ramion wykazują znaczne krzywizny - deformacje typu szpotawego, gdy dolne kończyny dziecka są zakrzywione w kształcie „koła” lub koślawe (w kształcie litery „X”). Kości klatki piersiowej i kręgosłupa są również zgięte.
- Opóźnienie w rozwoju psychicznym i fizycznym.
- Nietowarzystwo, nieśmiałość, złożoność.
- Polidypsja i wielomocz mogą postępować i ustępować, nie ustępując całkowicie.
- Umiarkowana hipotonia mięśni, wyrażająca się spowolnieniem, trudnościami w ruchach, prowadząca do tego, że dzieci w wieku 5–6 lat nie są w stanie chodzić.
- Bóle kości, o umiarkowanym natężeniu, uniemożliwiające dziecku chodzenie. Są one bardziej widoczne na poziomie nóg, miednicy i kręgosłupa. Chód, jeśli dziecko chodzi, staje się „kaczy” i niepewny.
- Wysokie prawdopodobieństwo złamań kości rurkowych z powodu niedoboru minerałów w tkance kostnej.
- Osteomalacja czyli rozmiękanie kości na skutek zniszczenia tkanki kostnej w wyniku braku soli wapnia i fosforu.
- Zmniejszona obrona immunologiczna przed infekcjami, która objawia się częstymi chorobami wirusowymi, zapaleniem ucha środkowego, zapaleniem płuc.
- Paraliż spowodowany brakiem potasu.
- Patologie okulistyczne, takie jak barwnikowe zwyrodnienie siatkówki, wrodzona zaćma.
- Rozwój patologii układu nerwowego, laryngologicznego (ucha, nosa, gardła, krtani) i przewodu pokarmowego, układu sercowo-naczyniowego, nieprawidłowości anatomiczne układu moczowego na skutek masywnych zaburzeń metabolicznych.
- W pojedynczych przypadkach - zaburzenia endokrynologiczne
Wraz z postępem zaburzeń kanalikowych (upośledzony transport substancji organicznych, minerałów, elektrolitów) w wieku 10–12 lat, u dzieci wzrasta ryzyko rozwoju przewlekłej niewydolności nerek, która zagraża ich życiu.
Widoczne objawy zespołu Fanconiego u dzieci 
U dorosłych pacjentów z rozwojem postaci wtórnej
Jeśli nabyty zespół Fanconiego rozwija się u dorosłych z innymi chorobami lub stanami patologicznymi, wówczas jego objawy często łączą się z objawami choroby wywołującej.
Zidentyfikowano jednak podstawowe objawy:
- Zwiększona objętość moczu na dzień (do 2 litrów lub więcej) i ostre pragnienie, również charakterystyczne dla pacjentów we wczesnym dzieciństwie.
- Osłabienie ogólne i mięśniowe, ból kości.
- Wysokie prawdopodobieństwo trwałego wzrostu ciśnienia krwi () z powodu dysfunkcji nerek.
- Osteomalacja (zniszczenie kości).
- Kwasica (zwiększona kwasowość krwi) spowodowana zatrzymywaniem produktów utleniania w organizmie, prowadząca do hipokaliemii (niedoboru potasu).
- Nefrokalcynoza to charakterystyczne dla tej choroby duże odkładanie się soli wapnia w nerkach z gorączką, dreszczami, nudnościami, silnym bólem brzucha, pachwin i jajników.
- Hipokaliemia (niskie spożycie potasu), która powoduje poważne powikłania kardiologiczne, w tym zagrażające życiu zaburzenia rytmu.
- Szybki (w przypadku braku leczenia) rozwój przewlekłej niewydolności nerek
Wśród kobiet
Najbardziej niekorzystny przebieg zespołu Fanconiego występuje, gdy rozwija się on u kobiet po menopauzie. W tym czasie, na tle zmniejszonej produkcji hormonów, następuje naturalny spadek gęstości kości (osteopenia).
Kiedy ten stan łączy się ze zwiększoną łamliwością kości spowodowaną brakiem minerałów, istnieje duże prawdopodobieństwo poważnych złamań kompresyjnych kręgów, szyjki kości udowej i późniejszej niepełnosprawności.
Diagnostyka
Aby potwierdzić lub obalić diagnozę, wykonuje się badanie kości oraz szczegółowe badania biochemiczne krwi i moczu za pomocą promieni rentgenowskich.
Laboratorium
Wykrywalne zmiany w biochemii moczu i krwi:
| Oznaki | Wskaźniki |
|---|---|
| niska zawartość wapnia i fosforu | odpowiednio poniżej 2,1 mmol/l i 0,9 mmol/l |
| kwasica („zakwaszenie” krwi) | BE = 10 – 12 mmol/l |
| cukromocz (zwiększony poziom cukru w moczu) | 2 – 3% i więcej |
| hiperaminocyduria (wydalanie ważnych aminokwasów alaniny, argininy, glicyny, prolitu z moczem) | do 2 – 2,5 g/dzień |
| wydalanie wapnia z moczem | 1,5 – 3,5 mmol/dzień |
| zwiększone pH moczu (kwasowość) z powodu nienormalnie dużej utraty wodorowęglanów | do 6,0 |
| wzrost względnej gęstości moczu | 1,025 – 1,035 |
Badanie ujawnia również:
- zwiększona aktywność fosfatazy alkalicznej;
- nadmierne wydalanie soli sodowych i potasowych z organizmu;
- białkomocz (pojawienie się białka w moczu) w obecności łańcuchów lekkich immunoglobulin, lizozymu, białek o niskiej masie cząsteczkowej, beta 2-mikroglobulin;
- zwiększony klirens (szybkość filtracji) kwasu moczowego przy zmniejszonej jego zawartości w surowicy
- zmniejszenie aktywności enzymów wymiany energii: dehydrogenazy bursztynianowej, dehydrogenazy a-glicerofosforanowej, dehydrogenazy glutaminianowej;
- zwiększenie ilości kwasu mlekowego i pirogronowego we krwi.
Instrumentalny
Rozpoznanie zespołu Fanconiego wymaga obowiązkowego wykonania radiografii kości w celu wykrycia deformacji szkieletu, kończyn, wykrycia objawów osteomalacji, osteoporozy, a u dzieci dodatkowo opóźnionego wzrostu kości w porównaniu z normą dla wieku kalendarzowego.
Obserwuje się następujące anomalie w tkance kostnej:
- gruboziarnista, komórkowa struktura z lukami, słaba mineralizacja, nieprawidłowe narośla w postaci „kolców” w kości udowej i piszczelowej;
- objawy epifizjolizy (częściowe lub całkowite ustanie wzrostu kości na długość w niedojrzałym szkielecie, prowadzące do asymetrii kończyn);
- złamania kości rurkowych (w późnym stadium) na tle rozwoju osteoporozy, którego stopień określa się za pomocą densytometrii rentgenowskiej;
- gromadzenie się radioizotopu w obszarach intensywnego wzrostu kości.
W nerkach:
Elektroniczne badanie biopsji tkanki nerkowej (biopsja) ujawnia charakterystyczną zmianę kształtu kanalików w postaci „łabędziej szyi”, przerzedzenie, zanik (zmniejszenie objętości) tkanki nabłonkowej w obecności zwiększonej liczby znajdujących się w nim mitochondriów, zwłóknienie (nieprawidłowy wzrost) tkanki łącznej.
Wymagane badania:
Mechanizm różnicowy
Patologię należy odróżnić od wszystkich izolowanych chorób, które mogą powodować wystąpienie nabytego zespołu Fanconiego, nabytych niezdrowych stanów i zatruć.
Ponadto u niemowląt pediatra musi różnicować stan ostrego niedoboru witaminy D w zespole Fanconiego od jej nadmiaru spowodowanego stosowaniem sztucznych suplementów lub zaburzeniami metabolizmu wapnia.
Różnice w hiperwitaminozie D i zespole de Fanconiego u dzieci poniżej pierwszego roku życia:
| Opcje | Hiperwitaminoza D | Zespół De Fanconiego |
|---|---|---|
| Częstotliwość | Często | Rzadko |
| Objawy (podobne) | Suchość skóry, bladość, ostre pragnienie, wymioty, długotrwałe zaparcia, niedobór masy ciała i wzrostu, powiększenie wątroby | |
| Objawy (różne) | Nadciśnienie (częste wzrosty ciśnienia krwi) | Wyczerpanie, wielomocz, hipotonia mięśni, brak wysokiego ciśnienia krwi |
| Krew | Nadmiar wapnia w ostrym okresie. Obniżona zawartość fosforu. Fosfataza alkaliczna, cukier, białko - w normie | Wapń jest często w normie (może być niski). Zmniejsza się poziom glukozy i białka. Fosfor jest znacznie zmniejszony. Aktywność fosfatazy alkalicznej gwałtownie wzrasta - 2-3 razy. Zwiększona kwasowość krwi |
| Mocz | Próba wydalania wapnia Sulkowicza jest dodatnia. Obecność białka, krwi (więcej niż 2 - 3 czerwonych krwinek w polu widzenia), leukocytów. Cukier azotowo-aminowy jest zwykle normalny | Próba Sulkowicza jest ujemna. Zwiększone stężenie białka, fosforanów, cukru, aminoacyduria |
| Kości | Strefy zwapnień ulegają rozszerzeniu i zagęszczeniu | Osteoporoza kości długich, niedobór wapnia w obszarach zwapnień |
Specjaliści zajmujący się zespołem Fanconiego i jego powikłaniami: nefrolog, ortopeda, hematolog, endokrynolog, urolog, okulista, genetyk.
Leczenie
Racjonalna, dobrze zaprojektowana terapia może zmniejszyć wpływ na mózg, układ kostny i narządy nadmiernych strat niezbędnych aminokwasów, minerałów, glukozy i białek wydalanych z moczem.
Dlatego leczenie zespołu ma na celu następujące zadania:
- Maksymalna możliwa korekta niedoboru potasu, niedoboru wodorowęglanów, zmiany równowagi kwasowo-zasadowej krwi w celu zmniejszenia kwasicy.
- Terapia cukrzycy fosforanowej (krzywicy D-odpornej) ze szczególnym uwzględnieniem niedopuszczalności ograniczenia płynów.
- Leczenie choroby podstawowej, która wywołuje rozwój zespołu nabytego u dorosłych.
Lek
 Aby złagodzić utratę fosforu i wapnia, stosuje się specjalne preparaty z witaminą D, są to l,25(OH)D3 i l(OH)D3.
Aby złagodzić utratę fosforu i wapnia, stosuje się specjalne preparaty z witaminą D, są to l,25(OH)D3 i l(OH)D3.
Początkowe dawki witaminy D3 na dzień wynoszą 10 – 15 tys. IU. Dawkę zwiększa się stopniowo, zwiększając ją co 12 do 14 dni (pod kontrolą testu Sulkovicha i poziomu fosforu we krwi). W przypadku braku objawów zatrucia i niewielkiego wydalania wapnia z moczem, można zwiększyć dawkę, doprowadzając ją do 100–150 tys. IU dziennie i kontynuować terapię do normalnego poziomu fosforu i fosfatazy zasadowej we krwi zostają osiągnięte. Gdy ich wartości ustabilizują się, nie należy dalej zwiększać dawki.
Terapia witaminą D prowadzona jest w kilku cyklach, aby zapobiec kryzysom postępu deformacji krzywicowych w kościach.
Najlepszą opcją jest zastosowanie aktywnych metabolitów D3 – Oksidevit (0,5 – 1,5 mcg dziennie), kalciotriolu (Rocaltrol).
Uwzględnij suplementy wapnia (glukonian wapnia dziennie do 1,5 - 2 gramów), fosfor (0,5 - 1 gram dziennie), fitynę.
Z fosforanów nieorganicznych należy stosować mieszaninę Albrighta, biorąc 1 dużą łyżkę 4 do 5 razy dziennie. Fosforany stosuje się w postaci roztworu i tabletek w dawkach obliczonych na 10 mg na kilogram masy ciała, 4 razy dziennie (zawsze z preparatami witaminy D w celu uniknięcia nadczynności przytarczyc).
W przypadku ciężkiego niedoboru potasu stosuje się Panangin i Asparkam.
W przypadku każdej recepty stale monitoruje się CBS lub stan kwasowo-zasadowy krwi. Zwykle krew ma odczyn lekko zasadowy, a pH mieści się w zakresie 7,35 – 7,45. W kwasicy, gdy wartość pH spada poniżej 7,35, krew staje się silnie kwaśna.
W takich przypadkach należy podać wlew dożylny 4% roztworu wodorowęglanu sodu lub wypić roztwór (50–60 ml dziennie), który zawiera kwas cytrynowy – 2 gramy, cytrynian sodu – 3 g, cytrynian potasu – 3,3 g, wodę 100 ml, jest wskazany. 1 ml takiej alkalizującej mieszaniny zawiera 1 mmol sodu i potasu. Wysoką kwasowość krwi można również zneutralizować sodą oczyszczoną (wodorowęglanem sodu).
Niektórzy eksperci na podstawie praktycznych wyników zalecają Unitiol jako środek zwiększający aktywność enzymów zależnych od tiolu.
W przypadku cystynozy przepisuje się: Dithiotrental w dawce 25 mg na kilogram masy ciała pacjenta po 3 godzinach; Cysteamina w dawce dziennej 90 mg/kg.
Pozytywne rezultaty daje stosowanie penicylaminy, która obniża stężenie kwasu pirogronowego we krwi, zmniejsza stopień eliminacji aminokwasów i sprzyja wzrostowi rezerw zasadowych w organizmie.
Hormonalne anaboliki, w tym metylotestosteron, dobrze wpływają na funkcjonowanie kanalików nerkowych.
Leczenie szpitalne jest wskazane w przypadku oczywistych zaburzeń metabolicznych, w tym hipo- i hiperglikemii oraz deformacji układu kostnego.
Zapobieganie
Współczesna profilaktyka wrodzonego zespołu Fanconiego w przypadku wystąpienia podobnej patologii w rodzinie polega na wstępnej konsultacji genetycznej. Ryzyko zachorowania na tę chorobę u tzw. rodzeństwa, czyli sióstr i braci, wynosi około 25%.
W wtórnej postaci zespołu jego objawy zmniejszają się lub całkowicie zanikają po aktywnym leczeniu choroby podstawowej lub nabytego stanu patologicznego.
Prognoza
Rokowanie w chorobie zależy od postaci (pierwotnej, wtórnej) zespołu, nasilenia objawów i rozpoczęcia leczenia.
Na przykład objawy zespołu nabytego znikają po wyeliminowaniu przyczyny wywołującej.
W przypadku patologii wrodzonej, przy braku złogów cystyny w tkankach, przebieg choroby nie stwarza poważnego zagrożenia życia, w przeciwnym razie, zwłaszcza bez odpowiedniego leczenia, śmierć pacjenta przewidywana jest nawet na 10–20 lat od momentu wzrośnięcia niewydolność nerek.
Jednak nawet przy ciężkich zmianach w nerkach: odmiedniczkowym zapaleniu nerek, cewkowo-śródmiąższowym zapaleniu nerek, niewydolności nerek, z wczesną interwencją lekową w procesie patologicznym i długoterminową terapią, ustala się pewna równowaga w homeostazie, w której rokowanie dla normalnej jakości życia od kilkudziesięciu lat jest dobrze.
W praktyce lekarskiej znane są przypadki, gdy u dzieci w wieku 7–8 lat dziedziczny zespół Fanconiego został praktycznie „zatrzymany” wraz z pojawieniem się długotrwałej remisji, wyraźną poprawą stanu dziecka, a nawet wyzdrowieniem.
Jeśli funkcja nerek odpowiedzialna za wchłanianie zwrotne składników odżywczych jest upośledzona, wykrywa się zespół Fanconiego. Drugą nazwą tego schorzenia jest zespół de Toniego-Debreu-Fanconiego. W patologii obserwuje się poziom glukozy i białka oraz występują zaburzenia metaboliczne. Choroba jest dziedziczona. Rozpoznanie stawia się na podstawie wyników klinicznych. Celem leczenia jest wyrównanie stanów niedoborowych, eliminacja niewydolności nerek.
U dzieci zespół de Toniego-Debreu-Fanconiego powoduje krzywicę, upośledzenie fizyczne i osłabienie tkanki mięśniowej.
Opis i uzasadnienie
Choroba De Toniego-Debreu-Fanconiego objawia się ciężką dysfunkcją kanalików, w wyniku czego zostaje zakłócony proces wchłaniania zwrotnego substancji i jonów korzystnych dla organizmu. Jednocześnie zwiększa się uwalnianie wodorowęglanów i wykrywane są ogólne zaburzenia metaboliczne. Osobliwości:
- spadek zawartości wapnia i fosforu w osoczu krwi;
- zaburzenie równowagi kwasowo-zasadowej, które objawia się niską kwasowością i wodorowęglanami we krwi;
- normalne wydalanie wapnia w postaci końcowego metabolitu z moczem przy zwiększonym tempie naturalnego oczyszczania organizmu z fosforanów;
- patologicznie wysoka aktywność fosfatazy zasadowej;
- pojawienie się cukru w moczu;
- ogólne zakłócenie produkcji i wydalania produktów syntezy aminokwasów;
- zwiększona objętość wydalanego moczu o zmniejszonej gęstości.
 Choroba De Toniego-Debreu-Fanconiego u dzieci.
Choroba De Toniego-Debreu-Fanconiego u dzieci. Zespół Fanconiego rozwija się zarówno u dzieci, jak i dorosłych. Istnieją wrodzone i nabyte typy patologii. W zależności od nasilenia objawów oraz rodzaju i nasilenia zaburzeń metabolicznych wyróżnia się 2 postacie choroby:
- Pierwszy typ objawia się wyraźnym opóźnieniem w rozwoju fizycznym z oczywistą deformacją kości szkieletowych, ciężkim obrazem klinicznym, częstymi złamaniami na tle ostrego braku wapnia z powodu słabego wchłaniania substancji w jelicie.
- Druga opcja objawia się umiarkowanym stopniem opóźnienia rozwoju fizycznego, łagodnymi objawami z łagodnym zniszczeniem kości i wystarczającym poziomem wapnia.
Przyczyny choroby
Czynniki prowokujące dzielą się na te, które powodują nabytą postać zespołu de Toniego-Debreu-Fanconiego i te, które są odpowiedzialne za pojawienie się wrodzonej patologii. Następujące stany są uznawane za prowokatory zaburzenia wtórnego:
- nietolerancja fruktozy;
- brak enzymów komórkowych;
- przewlekłe zatrucie toksynami;
- ostry niedobór witaminy D.
Sam zespół jest spowodowany jedną złą dziedzicznością lub świeżą mutacją genu odpowiedzialnego za procesy metaboliczne. Zespół De Toniego-Debreu-Fanconiego często rozwija się nie sam, ale razem z cystynozą, galaktozemią, zespołem Wilsona, pierwotną tyrozynemią i nietolerancją fruktozy. Ta forma jest częściej rejestrowana u dzieci niż u dorosłych.
Czynniki prowokujące
 Ostry niedobór witaminy D może wywołać chorobę.
Ostry niedobór witaminy D może wywołać chorobę. Zespół Fanconiego występuje częściej u osób z następującymi chorobami dziedzicznymi:
- niepowodzenie procesów metabolicznych cystyny (aminokwasy w białkach);
- nietolerancja produktów mlecznych (nawet mleka matki);
- defekty różnych enzymów odpowiedzialnych za syntezę i rozkład glikogenu;
- niepowodzenie w metabolizmie aminokwasów aromatycznych;
- nietolerancja fruktozy;
- problemy z metabolizmem miedzi (choroba Konovalova-Wilsona);
- zaburzenie metabolizmu mieliny i dysfunkcja enzymu sulfatazy;
- ciągłe narażenie organizmu na toksyny (leki, trucizny, metale ciężkie);
- niepowodzenie procesów metabolicznych z udziałem białka, co prowadzi do odkładania się określonego kompleksu - amyloidu;
- ostry niedobór witaminy D.
Wrodzone i nabyte
Zespół Fanconiego to ciężka choroba przypominająca krzywicę, która może mieć charakter pierwotny (wrodzony) lub wtórny (nabyty).
Wrodzona patologia de Toni-Debra-Fanconi jest dziedziczna, to znaczy przenoszona z rodziców na potomstwo. Często rozwija się wraz z innymi chorobami genetycznymi. Schorzenie charakteryzuje się ostrym zaburzeniem procesów metabolicznych, w wyniku czego wraz z dysfunkcją kanalików nerkowych rozwija się ślepota, zespół powiększenia wątroby oraz zespół z utrzymującym się spadkiem stężenia i aktywności tkankowej hormonów tarczycy. Mechanizm dziedziczenia zespołu zależy od rodzaju patologii, z jaką się rozwija.
Nabyty zespół de Toniego-Debreu-Fanconiego jest często wywoływany przez różne leki:
- leki chemioterapeutyczne stosowane w leczeniu raka;
- leki przeciwretrowirusowe;
- przestarzała tetracyklina.
Prowokatorami rozwoju wtórnych form patologii są skomplikowany przeszczep nerek, rak krwi i ciężkie stany niedoboru (głównie z powodu braku witaminy D). Jeśli choroba wrodzona rozwija się w macicy i jest rejestrowana głównie u dzieci, wówczas postać wtórna może również rozwinąć się u dorosłych.
Mechanizm rozwoju i przepływu
 Zaburzone jest wchłanianie zwrotne substancji w nerkach.
Zaburzone jest wchłanianie zwrotne substancji w nerkach. Zespół De Toniego-Debreu-Fanconiego spowodowany jest wrodzoną mutacją genu odpowiedzialnego za procesy metaboliczne, ale w ostatnim czasie zdarzały się przypadki transformacji spowodowanej innymi patologiami. Z powodu upośledzonego wchłaniania wielu przydatnych substancji i jonów rozwijają się poważne stany niedoboru:
- przy braku aminokwasów pojawia się ciężka dystrofia, spowalnia wzrost i rozwój fizyczny;
- po usunięciu fosforu i wodorowęglanów proces mineralizacji kończy się niepowodzeniem wraz z początkiem zniszczenia tkanki kostnej;
- z nagromadzeniem cukru w moczu - naruszenie regulacji metabolizmu węglowodanów;
- gdy potas jest wydalany z moczem - zanik mięśni, ciśnienie krwi spada do 80 mmHg. Sztuka. i poniżej;
- w przypadku zaburzeń metabolicznych na dużą skalę - zniszczenie tkanki nerkowej (zwężenie światła ze spłaszczeniem nabłonka i skróceniem kanalików, w których następuje wchłanianie zwrotne).
Objawy u dzieci i dorosłych
Pierwsze objawy wrodzonego zespołu de Toniego-Debreu-Fanconiego pojawiają się w pierwszym roku życia, rzadziej - od 1,5 roku. Dziecko często oddaje mocz, obserwuje się subfibraliczność (do 37-38°C), zaparcia i wymioty. W wieku 6 lat, jeśli nie jest leczone, dzieci nie mogą chodzić, a w wieku 12 lat ich nerki przestają działać, co może prowadzić do śmierci. Pacjenci są niekomunikatywni, skomplikowani i bojaźliwi. Funkcje psychiczne i myślenia są w normie. Konsekwencją tego zespołu są problemy z układem nerwowym i wzrokiem, rozwój przewlekłego niedoboru odporności, dysfunkcja narządów moczowo-płciowych i przewodu pokarmowego.
Objawy u dzieci rozwijają się najczęściej na skutek niedoboru fosforanów (cukrzyca typu fosforanowego) i objawiają się:
- niestabilność chodu;
- niski wzrost;
- niska mobilność;
- okrągłość nóg;
- skrzywienie kości szkieletowych (w szczególności kręgosłupa);
- ból kości, przez co dziecko nie chce chodzić;
- gwałtowny wzrost objętości wydalanego moczu;
- spadek ciężaru właściwego na tle jakościowej zmiany składu moczu;
- atonia mięśni;
- ból kości;
- choroba hipertoniczna;
- przewlekła niewydolność nerek (nieleczona);
- osteoporoza, osteopenia z powodu zmniejszenia gęstości kości.
Choroba De Toni Debra Fanconiego, powszechnie nazywana zespołem Fanconiego, wiąże się z upośledzoną funkcją nerek. Zakłócenia zachodzące w organizmie człowieka z powodu tej choroby prowadzą do powikłań. Poniżej znajduje się artykuł, którego informacje pomogą czytelnikowi zrozumieć charakterystykę choroby.
Co to jest choroba Fanconiego
 Nazwa choroby pochodzi od nazwiska szwajcarskiego lekarza Fanconiego, który na początku ubiegłego wieku jako pierwszy zbadał przyczyny i objawy choroby. Fanconi zbadał niskie dzieci chore na krzywicę, u których wystąpiły zmiany w moczu. Lekarz zebrał te objawy i połączył je w jedną patologię - zespół Fanconiego.
Nazwa choroby pochodzi od nazwiska szwajcarskiego lekarza Fanconiego, który na początku ubiegłego wieku jako pierwszy zbadał przyczyny i objawy choroby. Fanconi zbadał niskie dzieci chore na krzywicę, u których wystąpiły zmiany w moczu. Lekarz zebrał te objawy i połączył je w jedną patologię - zespół Fanconiego.
Po dwóch latach obserwacji pacjentów Tony dodał do istniejących jeszcze dwa czynniki patologiczne: spadek poziomu fosforanów we krwi poniżej ustalonej normy i hiperaminoacydurię.
Patologia jest dziedziczona. Podczas badania mocz pacjenta jest wypełniony nadmiarem cukru, białka i fosforanów. Metabolizm również ulega spowolnieniu i zatrzymuje się wchłanianie produktów nerkowych do krwi.
Na świecie na każde 350 tysięcy noworodków przypada 1 dziecko z opisanym powyżej zespołem. Warto zaznaczyć, że u dorosłych rzadko rozwija się ta wada.
Przyczyny tej choroby
Obecnie przyczyny zespołu Fanconiego mogą być inne. Lekarze sugerują, że rozwój choroby jest związany z niewyjaśnioną mutacją genu, podczas której zmieniają się funkcje enzymów, zmniejsza się zawartość fosforu we krwi i zaburzone zostaje funkcjonowanie aminokwasów. Niektórzy skłonni są do wniosku, że przyczyną patologii jest dysfunkcja białek kanalików nerkowych, która rozwija się na skutek defektów w strukturze mitochondriów.
Inne przyczyny zespołu:
- zatrucie truciznami i toksynami, które powstają w wyniku przedostania się metali ciężkich do organizmu;
- niedobór witaminy D powodujący defekty na poziomie genetycznym;
- zaburzenia wchłaniania enzymów komórkowych niezbędnych dla organizmu;
- odkładanie się amyloidu w tkankach na skutek zaburzeń metabolizmu białek;
- zaburzenie metaboliczne organizmu zwane cystynozą;
- tyrozynemia.
Wiele badań pokazuje również, że choroba ta jest jednym z powikłań krzywicy. Krzywica pojawia się z powodu niedoboru enzymów komórkowych, podczas gdy najbardziej przydatne i niezbędne substancje opuszczają organizm ludzki z moczem, zamieniając tkankę kostną w zmiękczoną masę.
W związku z powyższymi informacjami nie ustalono dokładnych przyczyn zespołu Fanconiego, dlatego w medycynie patologię tę nazywa się cukrzycą glukofosfaminową.
Objawy wskazujące na istniejącą patologię
Wyraźna manifestacja patologii jest zauważalna w dzieciństwie. Od chwili narodzin dziecka do pierwszego roku życia zaczynają pojawiać się pierwsze oznaki:
- zwiększona objętość wydalanego moczu -;
- kneblowanie;
- pojawienie się zaparć;
- podwyższona temperatura ciała;
- ciągłe pragnienie - polidypsja;
- słabe mięśnie;
- wzdęcia – gazy w jelitach;
- ostre bolesne odczucia w kościach, z którymi dziecko stale płacze, można pomylić.
Choroba De Toniego Debre Fanconiego u dzieci zmniejsza możliwości intelektualne. Według cech fizycznych dziecko pozostaje w tyle za rówieśnikami. Jest to spowodowane brakiem witamin wydalanych z organizmu dziecka z moczem. Kończyny dolne upodabniają się do litery „X” ze względu na krzywiznę. Narządy dziecka kurczą się w miarę rozwoju zaniku włókien mięśniowych. Wszystko kończy się na tym, że w wieku 5 lat dziecko nie jest już w stanie zrobić małego kroku.
Niewczesne leczenie prowadzi do pogorszenia czynności nerek w miarę starzenia się młodzieży. Występują również skutki uboczne choroby: pogorszenie wzroku, zwiększone napięcie serca i naczyń krwionośnych oraz uszkodzenie centralnego układu nerwowego.
Znane formy zidentyfikowanej choroby
Choroba dzieli się na 2 formy:
- Podstawową postacią jest dziedziczenie choroby przez dziecko.
- Forma wtórna występuje, gdy osoba zachoruje w wieku dorosłym.
W patologii dziedzicznej defekty występują w chromosomie X. Mutacja jest przekazywana w sposób recesywny i dominujący. Specjalistom trudno jest dokonać dokładnej prognozy. Chorobę można zdiagnozować jedynie podczas karmienia piersią. W inny sposób pierwotna postać tego zespołu nazywana jest „infantylną”.
Stopień mutacji chromosomu X wyjaśnia dalsze objawy. Chorobę można dokładnie zidentyfikować na podstawie 3 głównych wad:
- aminoacyduria.
Ważne jest, aby wiedzieć! Jeżeli zostaną wykryte tylko 2 składowe wyżej opisanych zaburzeń, diagnozę nazywa się niepełną.
Metody diagnozowania choroby
Zespół Fanconiego można wykryć głównie za pomocą promieni rentgenowskich i krwi. Rentgen pokazuje co następuje:
- opóźnienie wzrostu u dzieci;
- powolne zespolenie kończyn podczas złamań;
- rachiocampsis;
- deformacja kości;
- zanik kości długich;
- w strefach wzrostu kończyn kostnych tworzy się luźność;
- zmniejszenie gęstości kości, powodując ich łamliwość.
Badania krwi mogą zdiagnozować patologię na podstawie następujących czynników:
- nadmierna zawartość potasu;
- zwiększony poziom parathormonu;
- niskie stężenie fosforu i wapnia;
- zwiększona kwasowość w organizmie;
- wzrasta aktywność enzymu fosfatazy alkalicznej.
Analiza moczu wskazuje na zespół Fanconiego w przypadku:
- nadmiar soli fosforanowych w moczu;
- naturiuria;
- zwiększenie poziomu aminokwasów w moczu;
- zwiększony poziom glukozy.
Notatka! W niektórych przypadkach do diagnozy wykorzystuje się badanie radioizotopowe, nefbiopsję i biopsję tkanki kostnej.
Sposoby leczenia tego problemu
Lekarze poważnie traktują leczenie patologii. Terapia terapeutyczna odbywa się kompleksowo: za pomocą leków i zabiegów chirurgicznych. Często do tego dochodzi dieta. Podczas leczenia głównym zadaniem lekarza jest uzupełnienie niedoboru fosforu we krwi oraz uzupełnienie niedoboru potasu.
Bez diety aminokwasowej
Pacjent musi przestrzegać diety, aby zmniejszyć szybkość wydalania aminokwasów z organizmu z moczem. Dlatego w menu znajdą się następujące produkty:
- Ziemniak;
- kapusta;
- nabiał;
- owoce i soki z nich;
- rodzynki;
- Daktyle;
- suszone śliwki;
- suszone morele.
Lekarz może przepisać pacjentowi dodatkowe leki witaminowe bogate w witaminę D i potas.
Interwencja chirurgiczna
Pacjent trafia pod nóż chirurga w przypadku poważnych ubytków tkanki kostnej, przez co nie ma możliwości wykonywania prostych ruchów. Operację można przeprowadzić dopiero po rozpoczęciu fazy spokojnej i remisji utrzymującej się przez 1,5 roku. Aby złagodzić objawy patologii, pacjentowi zaleca się masaż i kąpiele igłami sosnowymi i solą morską.
Leczenie lekami
Aby przywrócić prawidłowy poziom wapnia i fosforu we krwi, pacjent oprócz diety przyjmuje leki przepisane przez lekarza. Równie ważne jest przyjmowanie leku, którego aktywnym składnikiem jest witamina D. Początkowo dawka wynosi 10 000 IU dziennie.
Z biegiem czasu dawka będzie wzrastać do 100 000 IU dziennie. Gdy badania wykażą prawidłowy poziom fosforu i wapnia, zaprzestaje się przyjmowania witaminy D. W leczeniu wtórnej postaci choroby stosuje się leki zawierające fitynę i wapń.
Zespół Fanconiego, znany również jako choroba Toniego-Debreu-Fanconiego, to schorzenie nerek, w którym upośledzone jest wchłanianie zwrotne zdecydowanej większości substancji, któremu towarzyszy cukromocz (cukier w moczu), aminoacyduria (białko), hiperfosfaturia (fosforany) ) i wyraźne zaburzenia metaboliczne w organizmie. . Ten stan u dzieci prowadzi do opóźnień rozwojowych, krzywicy i osłabienia mięśni.

W przypadku zespołu Fanconiego zaburzone jest wchłanianie zwrotne substancji w nerkach
Przyczyny tego stanu są różne i nie zostały jeszcze określone. Z reguły zespół Fanconiego wiąże się z nietolerancją, niedoborem wielu komórkowych układów enzymatycznych (na przykład karboksykinazą fosfoenolopirogronianową), przewlekłym zatruciem organizmu toksynami, niedoborem witaminy D.
Ze względu na różnice w koncepcjach ten sam stan można również nazwać „idiopatycznym nerkowym zespołem Fanconiego” i „krzywicą D-oporną”. Czasami używa się określenia „cukrzyca glukofosfaminowa” lub „karłowatość nerkowa z krzywicą D-odporną”.
Występuje stosunkowo rzadko, co częściowo wyjaśnia brak informacji na temat tego zespołu. Zespół ten występuje średnio u jednego na 350 000 dzieci.
Rodzaje zespołu Fanconiego

Z powodu niedoboru fosforanów golenie przyjmują kształt litery O
Wyróżnia się zespoły wrodzone (pierwotne, idiopatyczne) i nabyte.
Uważa się, że pierwotny zespół Fanconiego jest sprzężony z chromosomem X.
Może być dominujący lub recesywny, tj. dziedziczony na wiele różnych sposobów, przewidzenie obecności u potomstwa jest dość trudne.
Genetycznie uwarunkowany zespół Fanconiego może być całkowity lub niepełny, tj. czasami pojawiają się tylko 2 z 3 klasycznych objawów (glukozuria, aminoacyduria, hiperfosfaturia).

Pierwotny zespół Fanconiego jest chorobą genetyczną
Wtórne z reguły jest konsekwencją tyrozynemii typu 1. Dziedziczne patologie nerek przyczyniają się do wystąpienia tego zespołu. Zespół Fanconiego dość często rozwija się po narządzie (przy niskiej zgodności tkankowej).
Choroba Toniego-Debreu-Fanconiego może być konsekwencją zatrucia rtęcią, uranem, ołowiem lub kadmem. Czasami rozwija się u pracowników produkcji chemicznej, a mianowicie w kontakcie z toluenem, lizolem i kwasem maleinowym.
Czasami zespół Fanconiego rozwija się podczas leczenia lekami platynowymi, gentamycyną i przeterminowanymi lekami tetracyklinowymi.
Objawy zespołu Fanconiego
Galeria niektórych objawów
 Częste oddawanie moczu
Częste oddawanie moczu  Rachiocampsis
Rachiocampsis  Gorączka
Gorączka
U dzieci większość objawów jest zwykle związana z niedoborem fosforanów (podobnie jak w cukrzycy fosforanowej). Pojawia się chwiejny chód („kaczy”), niski wzrost i brak aktywności. Stopniowo tworzy się kształt litery O w nogach, a inne kości szkieletu (zwłaszcza kręgosłup) ulegają wygięciu. Z powodu bólu kości dziecko niewiele chodzi. Ryzyko złamań z powodu tkanki kostnej jest bardzo wysokie.
Dzieci dorastają nietowarzyskie i bojaźliwe - funkcje poznawcze (mentalne) nie cierpią, ale tworzą się kompleksy.

Zaparcia mogą być również związane z chorobą Toniego-Debreu-Fanconiego
Pierwsze oznaki choroby w postaci wrodzonej pojawiają się już w pierwszym roku życia (czasami od 1,5 roku).
Dziecko zaczyna często oddawać mocz, pojawia się (37-38 0C), rozwijają się zaparcia i mogą wystąpić wymioty.
Z biegiem czasu rodzice zauważają opisane powyżej objawy niedoboru fosforu. W wieku 6 lat dzieci zazwyczaj nie są już w stanie samodzielnie chodzić. W miarę rozwoju procesu do 12 roku życia powstaje on, co jest obarczone śmiercią.
Zaburzenia metaboliczne prowadzą do patologii układu nerwowego, problemów ze wzrokiem, wad w rozwoju układu moczowo-płciowego, chorób jelit i przewlekłych niedoborów odporności.
U dorosłych objawia się zespół wtórny (częste oddawanie moczu), ogólne osłabienie, niedociśnienie mięśni i ból kości. Niewydolność nerek rozwija się dość aktywnie i rozwija się nadciśnienie tętnicze.
Najgorsze jest to, że nabyty zespół Fanconiego dotyka kobiety po menopauzie. Oprócz naturalnego spadku gęstości masy kostnej (osteopenia, osteoporoza) dodaje się łamliwość kości wynikającą z niedoboru minerałów. Sytuacja ta kończy się złamaniami kompresyjnymi trzonów kręgów, złamaniami głowy kości udowej i niepełnosprawnością pacjenta.

Osteoporoza jest objawem pomocniczym w diagnostyce zespołu Fanconiego
Rozpoznanie zespołu Fanconiego
Zespół ten występuje rzadko, doświadczony lekarz może go podejrzewać na podstawie zdjęcia RTG kości oraz zaawansowanych parametrów biochemicznych krwi i moczu.

Pojawienie się ostróg wiąże się z naruszeniem procesu tworzenia tkanki kostnej
Zespół Fanconiego często łączy się z osteoporozą ogólnoustrojową (osteoporoza jest w tym przypadku objawem pomocniczym). Tkanka kostna ma gruboziarnistą strukturę włóknistą i często widoczne są ostrogi kostne („ostrogi”). Densytometria służy do określania gęstości kości.
Kości są słabo zmineralizowane, co stwierdzono na podstawie analizy biopsji (próbki tkanki).
W przypadku nerek badanie ujawnia zwyrodnienie kanalików nerkowych (jak „łabędzia szyja”), nabłonek ulega zniszczeniu, a struktury nerek zostają zastąpione tkanką łączną.
Jedyną rzeczą, która ratuje pacjentów, jest to, że warstwa kłębuszkowa jest zaangażowana w ten proces jako ostatnia i aż do samego końca choroby funkcja nerek jest w jakiś sposób realizowana.
Mikroskopia nabłonka kłębuszków wykazuje dużą liczbę mitochondriów w komórkach.
Leczenie choroby
Pierwotnego zespołu Fanconiego nie można wyleczyć, ponieważ mówimy o zmianach strukturalnych w nerkach, a także o utrzymujących się, genetycznie uwarunkowanych zaburzeniach metabolicznych. U takich pacjentów stale utrzymuje się poziom potasu, leczy się kwasicę kanalikową nerkową i inne zaburzenia metabolizmu wody i soli. Cukrzyca fosforanowa w tym przypadku podlega standardowemu leczeniu tej choroby.
Pacjenci powinni pić dużo płynów w ciągu dnia.
Galeria niektórych metod leczenia zespołu Fanconiego
 Zakraplacze
Zakraplacze  Witaminy
Witaminy  Kąpiele sosnowe
Kąpiele sosnowe  Masaż
Masaż
W niektórych przypadkach wtórny zespół Fanconiego jest uleczalny, zwłaszcza jeśli choroba lub stan powodujący tę patologię nerek zostanie skutecznie wyeliminowany.
Terapia lekowa
Aby zrekompensować zaburzenia w metabolizmie fosforu i wapnia, przepisuje się metabolity witamin z grupy D - 1(OH)D3 lub 1,25(OH)D3. Dawki witamin są dostosowywane od 10 000 IU do 100 000 IU dziennie. Dawkowanie dobiera się pod stałą laboratoryjną kontrolą poziomu fosforu i wapnia we krwi. Przepisywane są suplementy wapnia i fityna per os.
Terapia ta wykonywana jest okresowo, w formie kursów.
Terapia dietetyczna w zespole Fanconiego
Główną zasadą diety jest ograniczenie usuwania szeregu substancji z organizmu m.in. aminokwasy zawierające siarkę i fosfor. Dieta polega na ograniczaniu soli i szerokim stosowaniu pokarmów alkalizujących.
Konieczne jest wprowadzenie do diety dużej ilości soków owocowych i mleka (przy normalnej tolerancji)
 Soki owocowe
Soki owocowe  Ograniczanie soli
Ograniczanie soli  mleko
mleko  Suszone owoce
Suszone owoce
Jeśli zaburzenia układu mięśniowo-szkieletowego staną się krytyczne, zaleca się korekcję chirurgiczną.
Wczesne rozpoznanie zaburzeń metabolicznych, takich jak zespół Fanconiego, u dzieci pozwala uniknąć szybkiego kalectwa dziecka, u dorosłych zmniejsza ryzyko złamań i ucisku korzeni nerwowych, a także zwiększa jakość i długość życia. Gdy pojawią się pierwsze objawy choroby (wielomocz, zmiana koloru moczu, bóle kości i stawów), należy zgłosić się do lekarza.
Zespół Fanconiego to ogólnoustrojowe zaburzenie metaboliczne charakteryzujące się dysfunkcją nerek. Tracą zdolność do ponownego wchłaniania i zwracania do krwioobiegu aminokwasów, glukozy, soli wapnia i fosforu wydalanych z moczem. Jest to wrodzona lub nabyta wada organiczna. Jest to niezwykle rzadkie zjawisko, występujące z częstością 1 przypadku na 350 tys. noworodków. Dysfunkcja ta jednak niezwykle ciężko wpływa na stan organizmu.
Nazywa się ją także zespołem de-Tony’ego-Debreu-Fanconiego, cukrzycą fosforanową, dziedzicznym zespołem Fanconiego. Niektórzy naukowcy uważają, że ta patologia występuje, gdy gwałtownie zmniejszają się zapasy ATP (kwasu adenozynotrifosforowego) w komórkach. Inni wysuwają teorię, że krzywicowe deformacje kości powstają na skutek niedoboru fosforu, zaburzeń równowagi kwasowo-zasadowej lub obu tych czynników. Jeszcze inni uważają, że dysfunkcja nerek nie jest spowodowana chorobą biochemiczną, ale wadą strukturalną. Dokładny powód nie został jeszcze ustalony.
W przypadku tej patologii dotknięte są kanaliki proksymalne nerek. Stężenie związków wapnia i potasu w krwiobiegu utrzymuje się w granicach normy. Reakcja moczu jest zasadowa lub obojętna. Na skutek znacznych strat wodorowęglanów rozwija się kwasica. Jest to niebezpieczny stan, w którym równowaga kwasowo-zasadowa zostaje zakłócona, a organizm ulega „zakwaszeniu”. Kwasica jest zwykle spowodowana jakąkolwiek rozlaną chorobą nerek.
Wrodzona patologia może mieć różny stopień nasilenia. W przypadku 3 defektów biochemicznych rozpoznaje się całkowitą dysfunkcję nerek, a w przypadku 2 defektów rozpoznaje się niepełną dysfunkcję nerek. Niektórzy naukowcy uważają, że dziedziczny zespół Fanconiego jest niezależną chorobą przypominającą krzywicę. Jednak najczęściej towarzyszy innym wrodzonym patologiom. To może być:
- cystynoza (choroba związana z krystalizacją aminokwasu cystyny w tkankach);
- Choroba Wilsona-Kovalova (patologia spowodowana złogami miedzi w nerkach, wątrobie, mózgu);
- galaktozemia (zaburzona przemiana cukru prostego w glukozę);
- zespół Lowe’a (patologia objawiająca się opóźnieniem wzrostu, rozwojem umysłowym, zaćmą i jaskrą);
- tyrozynemia (bierność enzymu wątrobowego rozkładającego aminokwas tyrozynę);
- nietolerancja fruktozy.
Nabyta dysfunkcja, nazwana na cześć Fanconiego, może wystąpić z powodu następujących czynników:
- leki toksyczne dla nerek (aspiryna, gentamycyna, przeterminowana tetracyklina, leki przeciwwirusowe Cidofovir, dydanozyna, leki przeciwnowotworowe streptozocyna, ifosfamid itp.);
- zatrucie solami metali ciężkich i innymi agresywnymi związkami chemicznymi;
- ostry niedobór witaminy D;
- szpiczak mnogi (rak krwi);
- amyloidoza (zaburzenie metabolizmu białek);
- przeszczep nerki.
Objawy choroby u dzieci
Wrodzony zespół Fanconiego objawia się najwyraźniej u dzieci. Jej objawy dają się we znaki już w pierwszym roku życia dziecka. Ten:
- wielomocz (częste oddawanie moczu z dużą ilością płynu wydalanego z organizmu);
- polidypsja (patologicznie silne pragnienie);
- podniesiona temperatura;
- drgawki;
- częste wymioty bez wyraźnego powodu;
- uporczywe zaparcia;
- wysypka na skórze;
- obrzęk stawów;
- powiększenie nerek, węzłów chłonnych, śledziony;
- predyspozycja do infekcji.

Wskutek codziennego usuwania witamin, mikroelementów i glukozy z organizmu poprzez płyny, dziecko pozostaje opóźnione w rozwoju psychicznym i fizycznym. Kości nóg stają się wygięte, zwiotczałe, a następnie mięśnie całkowicie zanikają. Często dzieci tracą zdolność samodzielnego chodzenia. W wieku 12–13 lat rozwija się przewlekła niewydolność nerek. Czasami pogarsza się wzrok. Ponadto w przypadku zespołu Fanconiego objawy patologii mogą wskazywać na połączone wrodzone wady naczyń krwionośnych i serca, narządów trawiennych i układu moczowo-płciowego.
Objawy choroby u dorosłych
Najczęściej zauważane:
- wielomocz;
- hipostenuria (zmniejszona względna gęstość moczu);
- bolące kości;
- osteomalacja (zmiękczenie tkanki kostnej i utrata siły);
- słabe mięśnie;
- przyspieszony postęp nadciśnienia;
- przewlekła niewydolność nerek (jeśli nie ma leczenia).
Wielomocz w zespole Fanconiego u dorosłych nie jest wyraźny, pod tym względem znacznie różni się od nadmiernej ilości moczu w moczówce prostej. Najczęściej zespół ten staje się objawem szpiczaka mnogiego lub choroby Waldenströma (złośliwa zmiana układu krwiotwórczego). Oprócz umiarkowanej wielomoczu, upośledzona czynność nerek objawia się zmniejszeniem ich funkcji koncentracji i pojawieniem się białka w moczu.
To, czy zespół Fanconiego występuje, czy nie, można ocenić na podstawie takich charakterystycznych objawów, jak wysokie stężenie aminokwasów, glukozy i fosforanów w moczu. Zwiększone wydalanie glukozy z moczem w tej patologii jest spowodowane dysfunkcją nerek. Jednakże stężenie cukru na czczo i poziom hiperglikemii u tych pacjentów zwykle mieszczą się w granicach normy.
Wielomoczowi zawsze towarzyszy osłabienie mięśni, które jest najbardziej odczuwalne w kończynach. Występuje z powodu niedoboru potasu. Ponadto pacjentów prawie zawsze nękają bóle kości.

Objawy kwasicy u dorosłych i dzieci są takie same. Ten:
- utrata apetytu;
- bezprzyczynowe wymioty;
- zaparcie lub biegunka;
- kwaśny zapach wydobywający się ze skóry lub ust;
- obniżone ciśnienie krwi;
- ból głowy;
- duszność;
- bezsenność;
- skrajne wyczerpanie.
Jednak lekarze odróżniają kwasicę u dorosłych od kwasicy u dzieci. Uważa się, że u niemowląt jest to zawsze wrodzona patologia. U dorosłych może to być dalszy rozwój choroby wieku dziecięcego lub powikłanie nabyte w wyniku uszkodzenia nerek. Objawy kwasicy często łączą się z objawami śródmiąższowego zapalenia nerek i kamicy moczowej. Kamienie nerkowe powstają w wyniku dużej utraty wapnia z moczem. W ciężkich przypadkach wykrywa się objawy osteoporozy i osteomalacji.
Diagnoza patologii
Wykrywanie zespołu de-Tony-Debreu-Fanconiego rozpoczyna się od wywiadu, badania pacjenta i badań laboratoryjnych. Charakterystycznymi oznakami patologii biochemicznej formuły krwi jest niski poziom wapnia, fosforu, potasu i sodu, a także glukozy, alaniny, glicyny i kwasu glutaminowego. Ale w moczu występuje wysoki poziom fosforu i glukozy, białka i leukocytów.
Przy znacznej utracie wodorowęglanów z moczem i nadmiernym gromadzeniu się kwasów w organizmie rozpoznaje się kwasicę metaboliczną. Prawie u wszystkich pacjentów występuje podwyższony poziom kwasu pirogronowego i mlekowego we krwi. Wykrywa się także spadek aktywności enzymów metabolizmu energetycznego.
Podczas diagnostyki instrumentalnej obowiązkowe jest USG nerek i moczowodów. Nefrobiopsja (badanie miniaturowych próbek nerek) pozwala zobaczyć deformację kanalików proksymalnych, które są wydłużone jak łabędzia szyja. W późniejszych stadiach choroby wykrywa się zanik kłębuszków nerkowych.

Rentgen zdeformowanych kończyn dolnych pozwala wykryć zwyrodnienie tkanki kostnej. U dzieci często dochodzi do ukrytych złamań chrząstek nasadowych, przez co kości przestają rosnąć na długość i stają się asymetryczne. W kości piszczelowej wykryto nowe narośla przypominające ostrogi.
W późniejszych stadiach rozwoju zespołu rozpoznaje się osteoporozę. Istnieje duże ryzyko złamań kości rurkowych. Stopień zaawansowania osteoporozy określa się za pomocą densytometrii rentgenowskiej. Niską mineralizację tkanki kostnej stwierdza się poprzez badanie jej wycinków, uzyskanych także w drodze biopsji.
Główne kryteria diagnostyczne:
- Znaczący niedobór masy ciała i wzrostu dziecka.
- Słabość funkcji statyczno-motorycznych.
- Deformacje szkieletu przypominające krzywicę (degradacja struktury tkanki kostnej potwierdzana jest w badaniu RTG).
- Zaburzenia elektrolitowe.
W przypadku zespołu Fanconiego u dorosłych lekarze muszą przeprowadzić diagnostykę różnicową, aby wykluczyć podobne patologie. Ten:
- choroby dziedziczne (cystynoza, galaktozemia, choroba Wilsona-Kovalova, tyrozynemia itp.);
- przewlekłe odmiedniczkowe zapalenie nerek;
- cukrzyca;
- wtórna nadczynność przytarczyc;
- szpiczak mnogi;
- zatrucie narkotykami, agresywnymi chemikaliami;
- rozległe oparzenia.
Leczenie patologii
Idealnie jest, gdy zespół Fanconiego leczy genetyk lub hematolog, jednak nie w każdej placówce medycznej dostępni są tacy specjaliści. Najczęściej tą patologią zajmuje się nefrolog lub urolog. W przypadku zaobserwowania objawów nadczynności przytarczyc (nadmiernego wydzielania przytarczyc) konieczna jest konsultacja z endokrynologiem. Jeśli wzrok się pogorszy, należy udać się na badanie do okulisty.

Taktyki leczenia obejmują:
- Uzupełnianie niedoborów elektrolitów.
- Likwidacja zaburzeń równowagi kwasowo-zasadowej.
- Leczenie niewydolności nerek.
- Łagodzenie bolesnych objawów.
Do leczenia farmakologicznego stosuje się:
- Kalcytriol, Oksidevit i inne preparaty witaminy D;
- glukonian wapnia;
- Fityna, glicerofosforan wapnia, fosforan glinu;
- roztwory alkalizujące Bicitra, Polycitra;
- Indometacyna, metylotestosteron, hipotiazyd (w przypadku ciężkiego uszkodzenia kanalików bliższych);
- Panangin, Asparkam;
- Cystyna, Merkaptamina;
- antybiotyki;
- kortykosteroidy itp.
Ponieważ zespół Fanconiego jest przewlekły, leczenie prowadzi się przez długi czas, w długich kursach po obowiązkowych przerwach. Często można przywrócić metabolizm do normy, złagodzić ciężkość choroby i zapobiec niebezpiecznym powikłaniom. Ale niezwykle trudno jest całkowicie pozbyć się zespołu de Toniego-Debreu-Fanconiego, często nawraca.
Terapia tego zespołu ma na celu wyeliminowanie kwasicy, uzupełnienie potasu, wodorowęglanu wapnia, fosforanów i innych elektrolitów. Najważniejszą rolę odgrywa dieta terapeutyczna. Pij dużo płynów i zmniejsz ilość soli. Jedzenie powinno być spożywane w małych porcjach, ale często. Węglowodany należy ściśle dawkować, aby we krwi nie było nadmiaru glukozy.
Podobne artykuły
-
Naleśniki z kremem kefirowym z dziurkami
Cienkie naleśniki kefirowe, koronkowe i z dziurkami, to kolejny rodzaj tych pysznych smażonych produktów, którym warto się przyjrzeć. Już je przygotowaliśmy i też miały dziury, będą pewne różnice w przepisach, ale też sporo podobieństw. W jednym z...
-
Co jest potrzebne, aby dostać się do szkoły lotniczej?
Zawód pilota to jeden z zawodów popularnych, choć trudny do zdobycia. Osoby pragnące latać samolotami podlegają rygorystycznym wymaganiom i warunkom ich spełnienia. Ale nie ma rzeczy niemożliwych, a to oznacza, że warto zostać pilotem...
-
Zupa grochowa z wędzonym kurczakiem
Proste przepisy krok po kroku na przygotowanie pysznej zupy grochowej z wędzonym kurczakiem 2017-09-27 Olga Barkas Ocena przepisu 2684 Czas (min) Porcje (osoby) Na 100 gramów gotowego dania 9 gramów. 9 gr. Węglowodany 8 g....
-
Jak zrobić napój drożdżowy
Od wielu lat pamiętam, jak jako dziecko w przedszkolu sanatoryjnym, gdzie szczęśliwie trafiłam na jakiś czas (jak na sezon, jak do obozu pionierskiego), zawsze dostawaliśmy drożdże pij po drzemce..
-
Szaszłyk jagnięcy z grubym ogonem
Nadeszła wiosna, a już niedługo słoneczne, piękne dni zaproszą nas do spędzenia większej ilości czasu na świeżym powietrzu, w wesołym towarzystwie. A co w tym przypadku może być lepszego niż rumiany, aromatyczny kebab? Podpowiemy Ci kilka świetnych przepisów...
-
Co zrobić, jeśli ryba jest przesolona
Jeśli potrzebujesz przygotować danie z lekko solonego produktu? Kto może być zainteresowany takimi pytaniami? Dla jakiej kategorii ryb moczenie będzie najbardziej pomocne? Dlaczego jest to konieczne? Metody usuwania nadmiaru soli są odpowiednie dla ryb,...