Нунен синдром и подобные ему заболевания. Смотреть что такое "Синдром Нунан" в других словарях.
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава РФ, г. Москва Ключевые слова
: дети, синдром Нунан, диагностика.
Key words
: children, syndrome Noonan, diagnostics.
В статье описан синдром Нунан (синдром Ульриха – Нунан, тернероидный синдром с нормальным кариотипом) – редкая врожденная патология, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадические случаи. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского – Тернера у особей женского и мужского пола с нормальным кариотипом. Представлено клиническое наблюдение. Показаны сложности дифференциальнодиагностического поиска, недостаточная информированность клиницистов о данном синдроме и важность междисциплинарного подхода.
Исторические факты
Впервые о необычном синдроме упомянул О. Kobylinski в 1883 году (фото 1).
Старейший известный клинический случай синдрома Нунан, описан в 1883 году О. Kobylinski
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти пациентах со стенозом клапана легочной артерии, малым ростом, гипертелоризмом, умеренным снижением интеллекта, птозом, крипторхизмом и скелетными нарушениями. Доктор Нунан, практиковавшая как детский кардиолог в университете Айовы, заметила, что у детей с редким типом порока сердца – стенозом клапана легочной артерии – часто наблюдались типичные физические аномалии в виде низкого роста, крыловидной шеи, широко посаженных глаз и низко расположенных ушей. Мальчики и девочки поражались одинаково. Доктор Джон Опиц, бывший студент Нунан, первым ввел в употребление термин «синдромом Нунан» для характеристики состояния детей, у которых отмечались признаки, похожие на описанные Нунан. Позже Нунан написала статью «Гипертелоризм с фенотипом Тернера», и в 1971 году на симпозиуме сердечнососудистых заболеваний название «синдром Нунан» стало официально признанным .
Этиология и патогенез
Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью (рис. 1). Ген синдрома Нунан локализован на длинном плече хромосомы 12 . Не исключена генетическая гетерогенность синдрома. Описаны спорадические и семейные формы синдрома с аутосомно-доминантной формой наследования. В семейных случаях мутантный ген наследуется, как правило, от матери, так как из-за тяжелых пороков развития мочеполовой системы мужчины с этим заболеванием часто бесплодны. Большинство описанных случаев являются спорадическими, вызванными мутациями de novo.
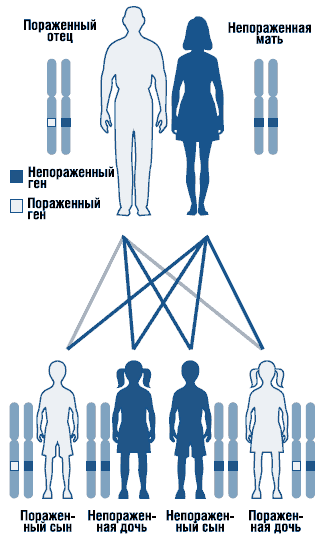
. Аутосомно-доминантный тип наследования
Описанные сочетания синдрома Нунан с нейрофиброматозом I типа в нескольких семьях заставило предположить возможную связь двух независимых локусов 17q11.2 хромосомы 17. У некоторых больных выявляются микроделеции в локусе 22q11 хромосомы 22; в этих случаях клинические проявления синдрома Нунан сочетаются с гипофункцией тимуса и синдромом Ди Джорджи. Ряд авторов обсуждают участие в патогенезе синдрома предполагаемых генов лимфогенеза в связи с наличием сходных с синдромом Тернера лицевых и соматических аномалий и высокой частоты патологии лимфатической системы .
Наиболее частая причина синдрома Нунан – мутация гена PTPN11, которая обнаруживается приблизительно у 50% больных. Белок, кодируемый геном PTPN11, относится к семейству молекул, регулирующих ответ эукариотических клеток на внешние сигналы. Наибольшее число мутаций при синдроме Нунан локализовано в экзонах 3,7 и 13 гена PTPN11, кодирующих домены белка, отвечающие за переход протеина в активное состояние .
Возможные представления о патогенезе представлены следующими механизмами:
RAS-MAPK-путь – очень важный путь сигнальной трансдукции, через который внеклеточные лиганды – определенные факторы роста, цитокины и гормоны – стимулируют клеточную пролиферацию, дифференцирование, выживаемость и метаболизм (рис. 2). После связывания лиганда рецепторы на поверхности клеток фосфорилируются в местах их эндоплазматического региона. Это связывание задействует адаптерные протеины (например, GRB2), которые формируют конститутивный комплекс с факторами обмена гуаниновых нуклеотидов (например, SOS), конвертирующих неактивный ГДФ-связанный RAS в его активную ГТФ-связанную форму. Активированные RAS-протеины затем активируют RAF-MEKERKкаскад через ряд реакций фосфорилирования. В результате активированный ERK проникает в ядро для изменения транскрипции целевых генов и корректирует активность эндоплазматических мишеней для индукции адекватных кратковременных и длительных клеточных ответов на стимул. Все гены, вовлеченные в синдром Нунан, кодируют интегральные для этого пути протеины, и мутации, вызывающие болезнь, обычно усиливают сигнал, проходящий через этот путь.
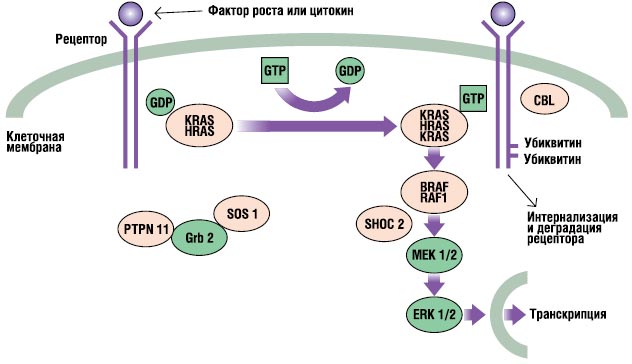
. RAS-MAPK-сигнальный путь. Ростовые сигналы передаются с активированных фактором роста рецепторов к ядру. Мутации в PTPN11, KRAS, SOS1, NRAS и RAF1 ассоциированы с синдромом Нунан, а мутации в SHOC2 и CBL ассоциированы с подобным синдрому Нунан фенотипом
Клиническая характеристика синдрома Нунан
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с крыловидной складкой или низким ростом волос, низкий рост, гипертелоризм глазных щелей (фото 2). Лицевые микроаномалии включают антимонголоидный разрез глазных щелей, опущенные вниз наружные углы глазных щелей, птоз, эпикантус, низко расположенные ушные раковины, складчатый завиток ушных раковин, аномалии прикуса, расщелину язычка мягкого неба, готическое небо, микрогнатию и микрогению. Грудная клетка щитовидной формы с гипоплазированными широко расставленными сосками, грудина выступает в верхней части и западает в нижней. Около 20% больных имеют умеренно выраженную патологию скелета. Наиболее часто встречаются воронкообразная деформация грудной клетки, кифоз, сколиоз; реже – уменьшение числа шейных позвонков и их сращение, напоминающее аномалии при синдроме Клиппеля – Фейля .
. Фенотипы синдрома Нунан
У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным ростом на темени, часто встречаются пигментные пятна на коже, гипертрихоз, дистрофия ногтевых пластинок, аномалии прорезывания и расположения зубов, склонность к образованию келоидных рубцов, повышенная растяжимость кожи. У трети больных отмечаются периферические лимфатические отеки, чаще лимфедема кистей и стоп проявляется у детей раннего возраста. Нередким признаком является патология зрения (миопия, косоглазие, умеренный экзофтальм и др.). Задержка роста встречается примерно у 75% больных, больше выражена у мальчиков и обычно незначительна. Отставание в росте манифестирует в первые годы жизни, реже отмечается незначительный дефицит роста и массы при рождении. С первых месяцев жизни отмечается снижение аппетита. Костный возраст обычно отстает от паспортного.
Характерным признаком синдрома является одно- или двусторонний крипторхизм, встречающийся у 70–75% больных мужского пола, у взрослых больных отмечается азооспермия, олигоспермия, дегенеративные изменения яичек. Тем не менее пубертат наступает спонтанно, иногда с некоторой задержкой. У девочек часто отмечается задержка становления менструации, иногда – нарушения менструального цикла. Фертильность может быть нормальной у больных обоих полов.
Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто отмечаются особенности поведения, расторможенность, синдром дефицита внимания. Речь обычно развита лучше, чем другие интеллектуальные сферы. Степень снижения интеллекта не коррелирует с тяжестью соматических нарушений [Маринчева Г.С., 1988]. В единичных случаях описываются пороки развития центральной нервной системы (гидроцефалия, спинномозговые грыжи), тромбоэмболические инфаркты мозга, возможно, связанные с гипоплазией сосудов .
Пороки внутренних органов при синдроме Нунан достаточно характерны. Наиболее типичными являются сердечно-сосудистые аномалии: клапанный стеноз легочной артерии (около 60% больных), гипертрофическая кардиомиопатия (20–30%), структурные аномалии митрального клапана, дефекты предсердной перегородки, тетрада Фалло; коарктация аорты описана только у больных мужского пола.
У трети больных регистрируются пороки мочевыделительной системы (гипоплазия почек, удвоение лоханок, гидронефроз, мегауретер и др.).
Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах в ротовой полости и носоглотке. Обнаруживаются различные дефекты коагуляции: недостаточность тромбоцитарной системы, снижение уровня факторов свертывания, особенно XI и XII, увеличение тромбопластинового времени . Имеются сообщения о сочетании синдрома Нунан с лейкемией и рабдомиосаркомой, что может свидетельствовать о некотором повышении риска малигнизации у этих больных .
В таблице 1 представлены особенности фенотипа при синдроме Нунан, меняющиеся с возрастом пациента. В таблице 2 – корреляция между фенотипом и генотипом при синдроме Нунан.
Таблица 1 . Типичные черты лица у больных синдромом Нунан по возрастам
| Лоб, лицо, волосы | Глаза | Уши | Нос | Рот | Шея | |
| Новорожденный* | Высокий лоб, низкая линия роста волос в затылочной области | Гипертелоризм, наклонные книзу глазные щели, складка эпикантуса | – | Короткий и широкий утопленный корень, вздернутый кончик | Глубоко утопленный губной желобок, высокие широкие пики красной каймы губ, микрогнатия | Избыточная кожа на затылке |
| Грудной (2–12 мес.) | Большая голова, высокий и выпирающий лоб | Гипертелоризм, птоз или толстые нависающие веки | – | Короткий и широкий утопленный корень | – | – |
| Ребенок (1–12 лет) | Грубые черты, вытянутое лицо | – | – | – | – | – |
| Подросток (12–18 лет) | Миопатичес-кое лицо | – | – | Мостик высокий и тонкий | – | Очевидное формирование шейных складок |
| Взрослый (>18 лет) | Отличительные черты лица утонченные, кожа кажется тонкой и прозрачной | – | – | Выпирающая носогубная складка | – | – |
| Все возрасты | – | Голубые и зеленые радужные оболочки, ромбовидные брови | Низкие, ротированные назад уши с толстыми складками | – | – | – |
Таблица 2 . Корреляции между генотипом и фенотипом при синдроме Нунан*
| Сердечнососудистая система | Рост | Развитие | Кожа и волосы | Другое | |
| PTPN11 (примерно 50%) | Более выражен стеноз легочного ствола; меньше – гипертрофическая кардиомиопатия и дефект межпредсердной перегородки | Более низкий рост; ниже концентрация IGF1 | Пациенты с N308D и N308S имеют слабое снижение или нормальный интеллект | – | Больше выражен геморрагический диатез и ювенильная миеломоноцитарная лейкемия |
| SOS1 (примерно 10%) | Меньше дефект межпредсердной перегородки | Более высокий рост | Меньше снижение интеллекта, задержка развития речи | Подобны сердечно-кожно-лицевому синдрому | – |
| RAF1 (примерно 10%) | Больше тяжелая гипертрофическая кардио-миопатия | – | – | Больше родимых пятен, лентиго, пятен кофе с молоком | – |
| KRAS (<2%) | – | – | Более тяжелая задержка когнитивного развития | Подобны сер-дечно-кожно-лицевому синдрому | – |
| NRAS (<1%) | – | – | – | – | – |
Данные лабораторных и функциональных исследований
Специфических биохимических маркеров для диагностики синдрома Нунан не существует. У некоторых больных выявляется снижение спонтанной ночной секреции гормона роста при нормальном ответе на фармакологические стимулирующие тесты (клофелином и аргинином), снижение уровня соматомедина-С и снижение реакции соматомединов на введение гормона роста.
Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях диагноз подтверждается результатами молекулярно-генетического исследования. Критерии диагностики синдрома включают наличие характерного лица (при нормальном кариотипе) в сочетании с одним из следующих признаков: патологии сердца, низкий рост или крипторхизм (у мальчиков), задержка полового созревания (у девочек). Для выявления сердечно-сосудистой патологии необходимо проведение ультразвукового исследования сердца с динамическим определением размеров полостей и стенки желудочков. Возможна пренатальная диагностика заболевания при помощи ультразвукового мониторинга, позволяющего выявить пороки сердца и аномалии строения шеи .
Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера; уточнить диагноз позволяет цитогенетическое исследование. Фенотипические признаки синдрома Нунан встречаются при ряде других заболеваний: синдроме Вильямса, синдроме LEOPARD, Дубовица, кардиофацио-кожном синдроме, Корнелии де Ланге, Коэна, Рубинштейна – Тейби и др. Точная идентификация этих заболеваний будет возможна только при проведении молекулярногенетических исследований каждого синдрома при значительном клиническом материале, что в настоящее время активно развивается.
Лечение
Лечение больных с синдромом Нунан направлено на устранение пороков сердечно-сосудистой системы, нормализацию психических функций, стимуляцию роста и полового развития. Для лечения больных с дисплазией клапанов легочной артерии, помимо прочих методов, с успехом применяется баллонная вальвулопластика. С целью стимуляции психического развития применяются ноотропные и сосудистые средства. Препараты, направленные на стимуляцию полового развития, показаны в основном больным с крипторхизмом. Применяются препараты хорионического гонадотропина в возрастных дозировках. В старшем возрасте – при наличии гипогонадизма – препараты тестостерона. В последние годы применяются рекомбинантные формы гормона роста человека в лечении больных с синдромом Нунан . Клинические данные подтверждаются увеличением на фоне терапии уровня соматомедина-С и специфического связывающего белка. Конечный рост больных, длительное время получающих терапию гормоном роста, в некоторых случаях превышает средний рост членов семьи.
Прогноз для жизни определяется тяжестью сердечно-сосудистой патологии.
Профилактика болезни основывается на данных медико-генетического консультирования.
Медико-генетическое консультирование
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в генах: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разрабатываются способы пренатальной диагностики заболевания.
Клиническое наблюдение
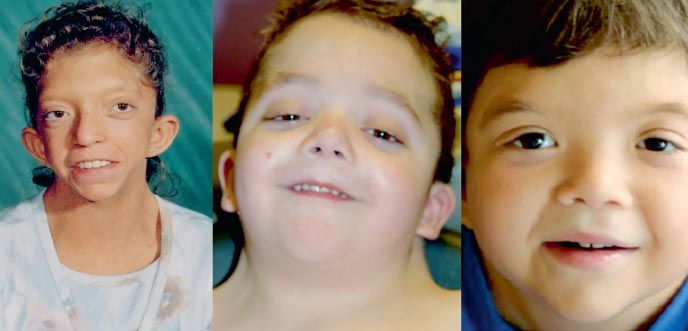
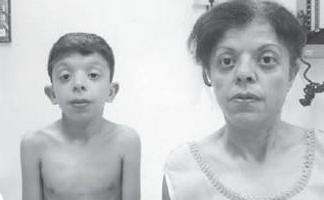
Мальчик Г., 9 лет (фото 3), наблюдался по месту жительства врачом-генетиком с диагнозом «хромосомная патология?, синдром Вильямса (своеобразный фенотип, уплотнение створок митрального клапана, гиперкальциемия однократно в 3 года)?.
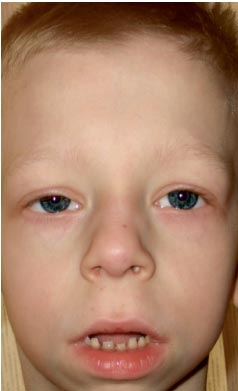
. Особенности фенотипа ребенка с синдромом Нунан (удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, нос укорочен с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия)
Жалобы на сниженную память, утомляемость, сниженные темпы роста.
Анамнез семейный : родители русские по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей, здоровые. Рост отца – 192 см, рост матери – 172 см. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии не отмечалось.
Анамнез жизни и заболевания : мальчик от 2-й беременности (1-я беременность – м/а), протекавшей с угрозой прерывания на всем протяжении, сопровождающейся многоводием. Роды первые, в срок, стремительные, масса при рождении – 3400 г, длина – 50 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. При рождении неонатологом обращено внимание на необычный фенотип ребенка, рекомендовано исследование кариотипа, результат – 46, XY (нормальный мужской кариотип). Был заподозрен врожденный гипотиреоз, проведено исследование тиреоидного профиля, результат – нормальный тиреоидный статус. Далее ребенок наблюдался генетиком с предполагаемым диагнозом «синдром Вильямса». Ранний постнатальный период – без особенностей. Моторное развитие по возрасту, первые слова – к году, фразовая речь – в 2 года 3 мес.
В возрасте 8 лет консультирован эндокринологом по поводу сниженных темпов роста, утомляемости, сниженной памяти. При рентгенологическом исследовании кистей рук выявлено умеренное отставание костного возраста (КВ) от паспортного (КВ соответствовал 6 годам). При исследовании тиреоидного профиля выявлено умеренное повышение тиреотропного гормона при нормальном уровне свободного Т4 и остальных показателей; УЗИ щитовидной железы – без патологии. Назначена гормональная терапия с последующим динамическим наблюдением.
Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Московский областной консультативно-диагностический центр для детей с целью уточнения диагноза.
Данные объективного исследования:
Рост – 126 см, вес – 21 кг.
Физическое развитие ниже среднего, гармоничное. Sds роста соответствует –1 (норма – –2+2). Особенности фенотипа (фото 3): удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, низкий рост волос на шее, нос укороченный с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия, гипертелоризм сосков, асимметрия грудной клетки, на стопах неполная кожная синдактилия 2–3-го пальцев, выраженная гипермобильность межфаланговых суставов, ломкие, сухие ногти. По внутренним органам – без особенностей. Половое развитие – Tanner I (что соответствует допубертатному периоду).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови – показатели в пределах нормы.
Тиреоидный профиль (ТТГ) – 7,5 мкМЕ/ мл (норма – 0,4–4,0), остальные показатели в норме.
Соматотропный гормон (СТГ) – 7 нг/мл (норма – 7–10), соматомедин-С – 250 нг/мл (норма – 88–360).
УЗИ щитовидной железы – без патологии.
УЗИ внутренних органов – без особенностей.
ЭКГ – синусовая тахикардия, нормальное положение электрической оси сердца.
ЭхоКГ – ПМК I степени с минимальной регургитацией, миксоматозное утолщение створок митрального клапана, дополнительная хорда в полости левого желудочка.
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника I степени.
R-графия кистей рук с захватом предплечий – костный возраст 7–8 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – без патологических изменений.
Аудиограмма – без патологии.
ДНК-диагностика: молекулярно-генетическое исследование – делеций исследуемых локусов критического района хромосомы 7 не выявлено; обнаружена мутация Gly434Ary (1230G>A) в 11-м экзоне гена SOS1 (анализ гена PTPN11 – мутаций не обнаружено), что характерно для синдрома Нунан.
Консультации специалистов:
Эндокринолог – субклинический гипотиреоз, неполная медикаментозная компенсация.
Окулист – астигматизм.
Невролог – вегетососудистая дистония. Невротические реакции.
Кардиолог – функциональная кардиопатия.
Хирург-ортопед – нарушение осанки. Деформация грудной клетки.
Генетик – синдром Нунан.
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований, поставлен диагноз «синдром Нунан», что подтверждено результатом молекулярно-генетического исследования.
Таким образом, представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний, важность молекулярно-генетических методов для уточнения диагноза. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждению повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии.
Список литературы:
- Baird P., De Jong B. Noonan’s syndrome (XX and XY Turner phenotype) in three generations of a family // J. Pediatr., 1972, vol. 80, p. 110–114.
- Hasegawa T., Ogata T. et al. Coarctation of the aorta and renal hupoplasia in a boy with Turner/Noonan surface anomalies and a 46, XY karyotype: a clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata // Hum. Genet., 1996, vol. 97, р. 564–567.
- Федотова Т.В., Кадникова В.А. и соавт. Клинико-молекулярно-генетический анализ синдрома Нунан. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика, приложение к № 5, 2010, с.184.
- Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? // Br. J. Dermatol., 1994, vol. 131, р. 270–274.
- Municchi G., Pasquino A.M. et al. Growth hormone treatment in Noonan syndrome: report of four cases who reached fi nal height // Horm. Res., 1995, vol. 44, р. 164–167.
Существуют различные врожденные патологии у детей. Некоторые из них считаются достаточно распространенными. Но есть и болезни редкие. Их распространенность невелика. Однако многие имеют достаточно серьезные последствия. Одной из таких патологий является синдром Нунан. Далее рассмотрим его более подробно. В статье будет описано, как проявляется и почему появляется синдром Нунан. Фото недуга также будет представлено в тексте.
Общие сведения
Синдром Нунан представляет собой наследственную патологию. Мутированный ген PTPN11 от носящих его родителей передается потомству. Как правило, мужчины при этом бесплодны. Поэтому ген передается по материнской линии. Обычно отмечается семейный характер этой болезни. Редкие случаи, не связанные с наследственностью, однако они также регистрируются в практике.
Историческая справка
В свое время практиковавшая как кардиолог-педиатр Жаклин Нунан, работая в клинике университета Айовы, заметила, что определенная группа детей, в которой состояли как мальчики, так и девочки, имевшие стеноз легочной артерии, часто отличалась низким ростом, расставленными широко глазами, перепончатой шеей, расположенными низко ушными раковинами и птозом. Исследовав сочетание проявлений порока сердца с прочими аномалиями развития у 833 пациентов, врач написала в 1962 году статью. В своей работе она описала девять случаев, при которых на фоне врожденного типа отмечались характерные черты лица, и низкорослость. Недуг обнаруживался как у лиц мужского, так и женского пола. 
Синдром Нунан: симптомы
Существует ряд характерных особенностей, отличающих патологию от прочих. К ним, в частности, относятся:
- Низкий рост. У женщин - 1,53, у мужчин - 1,63 м. На момент рождения как длина тела, так и вес ребенка находится в пределах нормы. Задержка в росте начинается с возраста 2-3 лет.
- Изменения лица. Пациенты, у которых диагностирован синдром Нунан, имеют расставленные широко формы. Во внутреннем углу присутствует кожная складка. Также отмечается птоз (опущены веки) из-за ослабленной мышечной функции либо вследствие мелких глазниц. Приблизительно две трети из всех детей с патологией имеют При птозе ограничивается и развивается косоглазие.
- Нарушения строения челюстей. Верхняя недоразвита, наблюдается арковидное высокое небо. На обеих челюстях взаиморасположение зубов неправильное.
- Низкая посадка либо деформация ушных раковин. В результате данной аномалии нарушен слух.
- Короткая и широкая шея.
- Щитообразная грудная клетка с расставленными широко сосками.
- Деформация в локтевом суставе (врожденная).
- Плоскостопие.
- Короткие пальцы.
Пороки развития внутренних органов
Синдром Нунан чаще всего сопровождается нарушениями в деятельности сердечно-сосудистой системы. На фоне патологии выявляется сужение (стеноз) в легочном стволе, а также дефект в межжелудочковой перегородке. Второе место занимают аномалии мочеполовой системы. Так, со стороны почек выявляются такие пороки, как гипоплазия (недостаточность развития ткани) или отсутствие одной почки. Что касается полового созревания, то оно варьируется от нормального до абсолютно дефектного. У девочек зачастую отмечается позднее наступление менструации, у мальчиков - крипторхизм либо яички полностью отсутствуют. Обнаруживаются также нарушения сперматогенеза. В ряде случаев сперматозоиды полностью отсутствуют. На фоне операций при синдроме Нунан отмечается повышенная кровоточивость. У некоторых пациентов также могут обнаруживаться отклонения в легкой степени. 
Формы
Существует два типа патологии:
- Семейная форма. Она отличается наследственным характером аутосомно-доминантного типа. У носителей мутантного гена потомство появляется с патологией.
- Спорадическая форма. В данном случае мутация появляется от случая к случаю. При этом наследственный фактор не выявлен.
Причины
Наиболее распространенным фактором, провоцирующим патологию, является мутация гена PTPN11. Эта причина выявляется у 50% пациентов. Тем не менее, у определенного процента людей с данным синдромом генетический фактор неясен. Наследование патологии происходит по аутосомно-доминантной форме. Синдром может быть спровоцирован новой мутацией. Из этого следует, что родители с таким геном не имеют больших шансов завести еще одного ребенка.
Диагноз
Его устанавливают в соответствии с характерными внешними признаками (они описаны выше). Также при диагностике исследуются лабораторные показатели. В частности, отмечается снижение концентрации тестостерона, XII фактора свертываемости. Проводятся также и инструментальные исследования. Назначается рентген грудины, эхокардиография, ЭКГ. С помощью этих методов выявляются аномалии внутренних органов. Врач может также назначить консультацию 
Терапевтические мероприятия
В связи с генетической обусловленностью синдрома лечение направлено главным образом на устранение симптомов. При крипторхизме назначается операция. В ходе нее яички перемещаются в мошонку. На фоне снижения концентрации андрогенов рекомендована гормональная терапия. При наличии почечной недостаточности назначается гемодиализ. С его помощью осуществляется удаление из организма продуктов обменных процессов посредством внепочечной очистки крови.
Нунен синдром (J.A. Noonan, амер. эндокринолог, родился в 1928 г.) - наследственное заболевание, характеризующееся низкорослостью и аномалиями соматического развития. Наиболее полно описан Ж. Нунен в 1963 г. Может быть спорадическим или семейным, наследуемым по аутосомно-рецессивному типу. Патологически измененные гены, обусловливающие Н. с., локализуются в аутосомах (см. Наследственные болезни ). Половой хроматин и кариотип (см. Хромосомы ) соответствуют полу больного. Заболевание выявляется как у мужчин, так и женщин.
Основными клиническими проявлениями Н. с. являются низкорослость, нижних конечностей, крыловидные складки кожи на шее, низкая граница роста волос на затылке, характерные дерматоглифические изменения, гипертелоризм, птоз, антимонголоидный разрез глаз, деформация или низкая посадка ушных овин, асимметрия лица, широкая переносица, что делает больных похожими друг на друга (рис .). Характерны короткая шея, врожденная деформация локтевого сустава (cubitus valgus), брахидактилия, высокое небо, патологический прикус , плоскостопие, изменение формы и структуры позвонков и другие аномалии, отмечаемые при Шерешевского - Тернера синдроме . Часто обнаруживаются стеноз легочного ствола, дефект межжелудочковой перегородки. Другие врожденные пороки развития сердечно-сосудистой системы (см. Пороки сердца врожденные ) отмечаются реже. Половое развитие при Н. с. варьирует от нормального до полной дисгенезии гонад (см. Гермафродитизм ). У девочек часто наблюдается позднее начало менструаций, у мальчиков - крипторхизм , гипогонадизм . При гистологическом исследовании материала, полученного при биопсии яичек, обнаруживают уменьшение или полное отсутствие герминативных клеток и гиперплазию клеток Лейдига. Другими проявлениями Н. с. являются гинекомастия и задержка умственного развития. В сыворотке крови больных содержание тестостерона, эстрогенов (см. Половые гормоны ) и гонадотропинов (см. Гипофизарные гормоны ) нормально или изменено соответственно степени выраженности дисгенезии гонад. Концентрация гормона роста в крови в норме. Кариотип 46ХХ или 46XV соответствует гонадному и паспортному полу.
Диагноз устанавливают на основании характерных клинических признаков и данных лабораторных исследований. Н. с. чаще всего дифференцируют с синдромом Шерешевского - Тернера (табл .).
Лечение при наличии признаков гипогонадизма заключается в заместительной терапии препаратами половых гормонов. Применение гормона роста неэффективно. По показаниям проводится хирургическая коррекция врожденных пороков развития, лечение психических нарушений, связанных с задержкой умственного развития (см. Эндокринные психические расстройства ). Прогноз в отношении жизни благоприятный. Особое внимание следует уделять трудоустройству: выбор специальности должен способствовать социальной адаптации больных с учетом их умственных способностей и физических недостатков.
Дифференциально-диагностические признаки синдромов Нунен и Шерешевского - Тернера
Клинический признак |
Синдром Нунен |
Синдром Шерешевского - Тернера |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Половое развитие |
Синдром Нунан характеризуется множественными врожденными пороками развития, типичными для больных с синдромом Шерешевского - Тернера, при нормальном кариотипе. Иногда синдром отождествляют с синдромом Бонневи - Ульриха, имеющим сходную клиническую картину. Однако при синдроме Бонневи - Ульриха отсутствует порок сердца, который очень часто наблюдается при синдроме Шерешевского - Тернера и синдроме Нунан. У всех больных можно обнаружить более или менее выраженные симптомы гипогонадизма, хотя отдельные авторы считают, что при синдроме Нунан у части мальчиков половые органы развиваются нормально. Этиология. Синдром наследуется аутосомнодоминантно с низкой пенетрантностью гена. Распространение синдрома достаточно велико (1:1000 - 1:5000), но нет четкого диагностического критерия в виде аномального кариотипа, как у больных с синдромом Шерешевского - Тернера. В связи с этим выявляются не все случаи заболевания. Гипоталамо-гипофизарно-гонадные взаимоотношения (патогенез) при синдроме Нунан практически не изучены. Имеется единственное сообщение об особенностях репродуктивной системы у больного , на основании которого трудно делать обобщения. Мы исследовали содержание тестостерона и гонадотропных гормонов в крови, а также функциональные резервы яичек у 8 мальчиков с синдромом Нунан. Уровень тестостерона у всех больных был ниже нормы. Сниженной оказалась и экскреция 17-КС с мочой. Эти данные указывают на гормональную недостаточность яичек при синдроме Нунан. Уровни тестостерона и гонадотропных гормонов в сыворотке крови и экскреция 17КС с мочой (М±m) у больных с синдромом Нунан
Сочетание недостаточной секреции тестостерона и высокого уровня ЛГ у 4 больных указывает на первичное поражение яичек, главным образом интерстициальных клеток. Однако введение ХГ в течение 3 дней в суточной дозе 1500 ЕД/м2 привело к значительному увеличению содержания тестостерона в крови у этих мальчиков. Kauschunsky с соавт. (1977) при введении гонадолиберина больному с синдромом Нунан отметили выраженное повышение секреции гонадотропинов, как у больных с первичным гипогонадизмом, и в то же время положительную реакцию яичек на ХГ. Ограниченность наблюдений затрудняет интерпретацию данных. Можно предположить, что у больных снижена чувствительность рецепторов клеток Лейдига к эндогенному гонадотропину, но резервы семенников сохранены, что выражается, в увеличении секреции тестостерона после стимуляций большими дозами ХГ. У больных с низким содержанием тестостерона и нормальным уровнем гонадотропных гормонов в крови патогенез тестикулярной недостаточности, вероятно, близок к патогенезу нормогонадотропного гипогонадизма. Theintz, Savagl (1982) описали 5 мальчиков 11,8 - 16,5 лет с синдромом Нунан. Все они имели явные признаки гипогонадизма и отставали в половом развитии. У 4 из них затем половое развитие нормализовалось, причем у двух спонтанно, а у двух - после лечения тестостерном. Лишь у одного больного лечение ХГ и тестостероном было неэффективным. Таким образом, у больных с синдромом Нунан, несмотря на общность клинической картины, природа гипогонадизма может быть различной. Недостаточность репродуктивной системы у них может быть следствием первичного дефекта гонад, дисфункции гипоталамогипофизарной системы и изменения чувствительности семенников к гонадотропной стимуляции. Клиническая картина напоминает клинику синдрома Шерешевского - Тернера. Приводим выписку из истории болезни одного из наблюдаемых нами больных. Сережа Л., 8 лет, поступил в детское эндокринологическое отделение больницы им. Раухфуса 05.03.82 г. с отставанием в физическом и умственном развитии. Родился от второй беременности, протекавшей с тяжелым токсикозом. На 7 - 8-й неделе беременности мать перенесла острую респираторную вирусную инфекцию. Первая беременность закончилась выкидышем. Мать страдает врожденным . Мальчик родился преждевременно (извлечен путем кесарева сечения). Масса тела 3500 г, длина 51 см. В периоде новорожденности состояние тяжелое. В 4месячном возрасте диагностирован порок сердца; мальчик отставал в физическом и умственном развитии с первого года. Отсутствие яичек в мошонке выявлено при рождении. При поступлении длина тела 124 см (ниже средней), масса 22 кг. Телосложение неправильное - широкая короткая шея, асимметричная грудная клетка, воронкообразная деформация грудины; высокое небо, гипертелоризм, деформация ушных раковин, дефекты пальцев рук, широко расставленные соски. Половой член развит удовлетворительно, мошонка очень маленькая, яички не пальпируются. Интеллект умеренно снижен. Кариотип 46XY. Рентгенограмма черепа без особенностей. Костный возраст соответствует 7 годам. В сыворотке крови ЛГ 4,33 мМЕ/мл, ФСГ 3,13 мМЕ/мл, тестостерон 0,56 нмоль/л (все показатели снижены). Экскреция 17КС 9,1 ммоль/сут. Функциональная проба с однократным введением ХГ отрицательная - тестостерон крови - 0,57 нмоль/л, 17-КС - 8,87 ммоль/сут; 3дневная проба положительная - тестостерон - 1,54 нмоль/л, 17КС - 10,15 ммоль/сут. На основании анамнеза, клинической картины и лабораторных данных поставлен диагноз синдрома Нунан. Положительная 3дневная проба позволила провести лечение ХГ. При отсутствии эффекта показано хирургическое лечение крипторхизма. «Нарушения полового развития у мальчиков», Внешний вид.
Формы
Причины
ДиагностикаДиагноз устанавливают на основании:
Лечение синдрома Нунан
Осложнения и последствия
Профилактика синдрома Нунан
Похожие статьи
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||