Sindromul Fanconi la copii și adulți. Sindromul Fanconi: simptome, diagnostic și metode de tratament Cum să tratați această problemă
Sindromul Fanconi (nume complet - de Toni-Debre-Fanconi) este o patologie congenitală care se exprimă într-o disfuncție gravă a tubilor renali proximali, și anume, o încălcare a absorbției secundare (absorbția în sânge) a substanțelor filtrate de rinichi. , ceea ce duce la glucozurie (creșterea zahărului în urină), fosfaturie (deteriorarea metabolismului fosforului și calciului), aminoacidurie (excreție crescută de aminoacizi în urină) și scăderea concentrației de bicarbonați care reglează aciditatea sângelui.
Sindromul De Toni-Debre-Fanconi
Sindromul Fanconi este o boală foarte rară, întâlnită în principal la copii, iar conform statisticilor medicale, frecvența acesteia corespunde unui copil bolnav la 350 de mii de nou-născuți de ambele sexe.
La adulți, se observă extrem de rar, dezvoltându-se pe fondul patologiilor dobândite. Cod patologic conform ICD-10: E72.O.
Cauze
Natura și cauzele defectului genetic al sindromului Fanconi nu sunt bine înțelese astăzi.
Se presupune că patologia se bazează fie pe defecte ale proteinelor de transport ale tubilor renali, fie pe o mutație genetică care distorsionează funcția enzimelor care reglează reabsorbția glucozei, aminoacizilor și fosforului.
Există dovezi din studiile defectelor punctiforme ale mitocondriilor, care conduc la funcționarea necorespunzătoare a tubilor rinichi.
Boala este, de asemenea, asociată cu intoleranță la fructoză, otrăvire cronică cu toxine (metale grele, ifosfamidă, aminoglicozide), deficit de vitamina D, amiloidoză, insuficiență a unui număr de enzime celulare (piruvat carboxilază, fosfoenolpiruvat carboxikinaza și altele), tirozofiemie, leucodicromafiemie, galactozemie, cistinoză, glicogenoză.
Potrivit altor experți, sindromul Fanconi poate fi o patologie izolată - și anume, una dintre formele severe de patologii asemănătoare rahitismului care sunt ereditare.
Studiile confirmă că, în sindromul Fanconi, metabolismul energetic celular cu participarea ATP (adenozin trifosfat) și transportul intercelular în tubii elementului principal al rinichiului, nefronul, este perturbat.
Datorită insuficienței funcțiilor enzimatice, apar pierderi de glucoză, fosfați, aminoacizi, iar tubulii rinichilor suferă de o lipsă de energie. În același timp, substanțele importante pleacă cu urina, ducând la modificări distrofice ale țesutului osos - rahitism.
Deoarece medicina nu a ajuns încă la o concluzie clară cu privire la cauzele sindromului Faconi, această afecțiune este denumită și prin alți termeni: „diabet glucofosfaminic”, „sindrom renal idiopatic Fanconi”, „rahitism rezistent la D”, „nanism renal cu Rahitism rezistent la D” , „Sindromul ereditar Fanconi”.
Forme și patogeneză
Există două tipuri de sindrom Fanconi:
- ereditar (congenital, idiopatic), care se referă la forma primară;
- dobândită, care este considerată o formă secundară a bolii.
Experții asociază specia ereditară (genetică) cu un defect al cromozomului X, care este moștenit într-o manieră dominantă și recesivă, astfel încât predicția genetică a manifestării sale la urmașii viitori nu este o sarcină ușoară. Dacă patologia aparține tipului congenital (forma primară), atunci este detectată la un copil în perioada toracică de până la un an. Prin urmare, forma primară se numește „sugar”.
Gradul de mutație genetică determină, de asemenea, severitatea sindromului în sine. Deci, sindromul Fanconi ereditar complet se dezvăluie în prezența a 3 defecte biochimice de bază, care includ glucozurie, aminoacidurie, fosfaturie, incomplete - cu două dintre ele.
De obicei, o patologie determinată genetic este însoțită de alte afecțiuni congenitale: cistinoză, sindroame Wilson, Dent, Lowe, intoleranță la fructoză, tirozinemie (incapacitatea organismului de a descompune eficient tirozina aminoacidului), galactozemie (disfuncție de transformare a zahărului în glucoză). ), acumularea excesivă de glicogen.
Dezvoltarea sindromului Fanconi
Sindromul dobândit (secundar), spre deosebire de cel congenital, nu este însoțit, ci este o consecință a patologiilor deja existente:
- tirozinemia tip I;
- cistinoză (metabolismul afectat al aminoacidului cistină cu afectare ulterioară a rinichilor);
- intoleranță la fructoză;
- boala Wilson-Konovalov;
- galactozemie;
- glicogenoză (acumulare anormală de glicogen în țesuturi și organe) tip XI;
- patologii ereditare ale rinichilor;
- (încălcarea metabolismului proteic care duce la scleroză, atrofie, disfuncție de organ);
- tubulointerstițial (nefrită cu afectare tisulară, tubuli renali);
- hiperparatiroidism (o boală endocrină în care conținutul normal de calciu și fosfor din sânge este perturbat);
- afecțiuni maligne: mielom, plămân, pancreas, plămân, boală a lanțului ușor,;
- arsuri profunde.
În plus, sindromul poate provoca astfel de condiții:
- transplant de organe cu compatibilitate tisulară scăzută;
- deficit de vitamina D;
- intoxicații cu uraniu, bismut, mercur, plumb, cadmiu;
- contact cu toluen, acid maleic, lizol;
- utilizarea agenților farmacologici nefrotoxici, cum ar fi: gentamicina, preparate de platină, medicamente pe bază de tetracicline expirate, Didanozină, Cidofovir, medicamente pentru chimioterapie anticancer - Ifosfamidă, Streptozocin.
Simptome și semne
Cu o formă ereditară (congenitală).
Simptomele primare apar în primele luni de viață, rar - după un an și jumătate.
În primul rând, la un nou-născut sunt observate următoarele condiții:
- urinare frecventă (poliurie);
- sete crescută (polidipsie);
- constipație prelungită;
- atacuri frecvente de vărsături fără cauză;
- astenie (oboseală generală), slăbiciune musculară;
- „sărituri” inexplicabile de temperatură până la 37,5 - 38 C;
- burtă umflată.
De regulă, în perioada în care încep crizele de vărsături și temperatura crește, copilul este prezentat medicului pediatru. Un specialist cu experiență ar trebui să stabilească că combinația de semne tulburătoare pentru părinți nu are legătură cu infecțiile respiratorii acute, infecțiile virale respiratorii acute sau infecțiile cu enterovirus.
Un medic pediatru competent este capabil să recunoască sindromul Fanconi la timp. Și teste de laborator - pentru a confirma suspiciunile dacă dezvăluie trei (sau două) semne de bază: glucozurie, hiperaminoacidurie generalizată și hiperfosfaturie, caracteristice acestei patologii.
După simptome ușoare și destul de vagi în următorul an și jumătate, simptomele caracteristice sindromului Fanconi sunt clar fixate:
- Nanism timpuriu (statură mică), cauzat de excreția constantă a celor mai importanți aminoacizi, glucoză, calciu, fosfați din organism. Primele șase luni de creștere și greutate normale sunt înlocuite de o deficiență a greutății corporale (până la 30%) și de creștere (de la 2 la 21%).
- Rahitismul, cauzat de excreția masivă de calciu și fosfați, devine vizibil după 10-12 luni de viață și are trăsături caracteristice sindromului Fanconi: capul copilului este de obicei ușor deformat, dar oasele mari ale picioarelor și brațelor prezintă o curbură semnificativă - deformări de tip varus, când tibiele bebelușului sunt îndoite de o „roată”, sau valgus (sub forma literei „X”). Oasele pieptului și ale coloanei vertebrale sunt, de asemenea, îndoite.
- Întârziere în dezvoltarea mentală și fizică.
- Lipsa de sociabilitate, frică, complexe.
- Polidipsia și poliuria pot progresa și regresa fără să dispară complet.
- Hipotensiune musculară moderată, exprimată prin lentoare, dificultăți în mișcări, ceea ce duce la faptul că copiii de 5-6 ani nu sunt capabili să meargă.
- Dureri la nivelul oaselor, de intensitate moderată, împiedicând copilul să meargă. Sunt mai pronunțate la nivelul picioarelor, bazinului și coloanei vertebrale. Mersul, dacă copilul merge, devine „rață”, nesigur.
- Probabilitate mare de fracturi ale oaselor tubulare din cauza unei deficiențe de minerale în țesutul osos.
- Osteomalacia sau înmuierea oaselor din cauza distrugerii țesutului osos ca urmare a lipsei de săruri de calciu și fosfor.
- Protecție imunitară redusă împotriva infecțiilor, care se manifestă prin boli virale frecvente, otită, pneumonie.
- Paralizie din cauza lipsei de potasiu.
- Patologii oftalmice, cum ar fi retinita pigmentară, cataracta congenitală.
- Dezvoltarea patologiilor sistemului nervos, ORL (ureche, nas, faringe, laringe) și tractului gastro-intestinal, sistemului cardiovascular, anomalii anatomice ale sistemului urinar din cauza tulburărilor metabolice masive.
- În cazuri izolate - tulburări endocrine
Odată cu progresia tulburărilor tubulare (transport afectat de substanțe organice, minerale, electroliți) până la vârsta de 10-12 ani, copiii sunt mai susceptibili de a dezvolta insuficiență renală cronică, care le amenință viața.
Simptome vizibile ale sindromului Fanconi la copii 
La pacienții adulți cu dezvoltarea unei forme secundare
Dacă sindromul Fanconi dobândit se dezvoltă la adulți cu alte boli sau stări patologice, atunci manifestările sale sunt adesea combinate cu manifestările bolii provocatoare.
Cu toate acestea, caracteristicile de bază sunt:
- Un volum crescut de urină pe zi (până la 2 litri sau mai mult) și sete acută, care sunt, de asemenea, caracteristice pacienților din prima copilărie.
- Slăbiciune generală și musculară, dureri osoase.
- Probabilitate mare de creștere persistentă a tensiunii arteriale () pe fondul disfuncției renale.
- Osteomalacia (defalcarea oaselor).
- Acidoza (creșterea acidității sângelui) datorată reținerii produselor de oxidare în organism, ducând la hipokaliemie (deficit de potasiu).
- Nefrocalcinoza este o depunere mare de săruri de calciu în rinichi cu febră, frisoane, greață, dureri severe la nivelul abdomenului, inghinal și ovare, care sunt caracteristice acestei afecțiuni.
- Hipokaliemie (aport scăzut de potasiu) care provoacă complicații cardiace severe, inclusiv aritmii care pun viața în pericol.
- Dezvoltarea rapidă (dacă netratată) a insuficienței renale cronice
Printre femei
Evoluția cea mai nefavorabilă a sindromului Fanconi apare atunci când se dezvoltă la femeile aflate în postmenopauză. În acest moment, pe fondul scăderii producției de hormoni, are loc o scădere naturală a densității osoase (osteopenie).
Când această afecțiune este combinată cu o creștere a fragilității osoase din cauza lipsei de minerale, probabilitatea unor fracturi severe de compresie ale vertebrelor, colului femural și dizabilității ulterioare este mare.
Diagnosticare
Pentru a confirma sau infirma diagnosticul, raze X sunt folosite pentru a examina oasele și studii biochimice aprofundate ale sângelui și urinei.
laborator
Modificări detectabile în biochimia urinei și sângelui:
| semne | Indicatori |
|---|---|
| conținut scăzut de calciu și fosfor | mai puțin de 2,1 mmol/l și, respectiv, 0,9 mmol/l |
| acidoză („acidificarea” sângelui) | BE = 10 - 12 mmol/l |
| glucozurie (creșterea zahărului în urină) | 2 - 3% și mai mult |
| hiperaminocidurie (excreția urinară a aminoacizilor importanți alanină, arginină, glicină, vărsare) | până la 2 - 2,5 g/zi |
| excreția de calciu în urină | 1,5 - 3,5 mmol / zi |
| o creștere a pH-ului (acidității) urinei datorită unei pierderi anormal de mare de bicarbonat | până la 6.0 |
| creșterea densității relative a urinei | 1,025 – 1,035 |
Examinarea mai relevă:
- activitate crescută a fosfatazei alcaline;
- excreția excesivă a sărurilor de sodiu și potasiu din organism;
- proteinurie (apariția proteinelor în urină) în prezența lanțurilor ușoare de imunoglobuline, lizozim, proteine cu greutate moleculară mică, beta 2-microglobuline;
- clearance-ul crescut (viteza de filtrare) a acidului uric cu conținutul său redus de ser
- o scădere a activității enzimelor de metabolism energetic: succinat dehidrogenază, a-glicerofosfat dehidrogenază, glutamat dehidrogenază;
- o creștere a cantității de acid lactic și piruvic din sânge.
instrumental
Diagnosticul sindromului Fanconi prevede utilizarea obligatorie a radiografiei osoase pentru a detecta deformarea scheletului, a membrelor, a detecta semnele de osteomalacie, osteoporoză, iar la copii - în plus - creșterea osoasă piertă în comparație cu norma pentru vârsta calendaristică.
Se observă următoarele anomalii în țesutul osos:
- fibros grosier, structură celulară cu lacune, mineralizare slabă, creșteri anormale sub formă de „spini” la femur și tibie;
- semne de epifizioliză (oprirea parțială sau completă a creșterii osoase în lungime în scheletul imatur, ceea ce duce la asimetria membrelor);
- fracturi ale oaselor tubulare (într-un stadiu târziu) pe fondul dezvoltării osteoporozei, al cărei grad este determinat cu ajutorul densitometriei cu raze X;
- acumularea unui radioizotop în zonele de creștere osoasă intensivă.
În rinichi:
Un studiu electronic al unei biopsii a țesutului renal (biopsie) relevă o modificare caracteristică a formei tubilor sub forma unui „gât de lebădă”, subțierea, atrofia (reducerea volumului) țesutului epitelial în prezența unui creșterea numărului de mitocondrii în ea, fibroză (creștere anormală) a țesutului conjunctiv.
Cercetare necesară:
diferenţial
Patologia ar trebui să fie distinsă de toate bolile izolate care pot provoca apariția sindromului Fanconi dobândit, stări nesănătoase dobândite și intoxicații.
În plus, la sugari, medicul pediatru este obligat să diferențieze starea de deficiență acută de vitamina D în sindromul Fanconi de excesul acestuia atunci când se utilizează suplimente artificiale sau tulburări ale metabolismului calciului.
Diferențele dintre hipervitaminoza D și sindromul de Fanconi la copiii sub un an:
| Opțiuni | Hipervitaminoza D | sindromul de Fanconi |
|---|---|---|
| Frecvență | De multe ori | Rareori |
| Simptome (asemănătoare) | Piele uscată, paloare, sete acută, vărsături, constipație prelungită, deficiență de greutate și înălțime, mărire a ficatului | |
| Simptome (variate) | Hipertensiune arterială (creșteri frecvente ale tensiunii arteriale) | Epuizare, poliurie, hipotensiune musculară, fără hipertensiune arterială |
| Sânge | Exces de calciu în perioada acută. Conținut redus de fosfor. Fosfataza alcalina, zahar, proteine - normal | Calciul este adesea normal (poate fi scăzut). Glucoza, proteinele sunt reduse. Fosforul este redus brusc. Activitatea fosfatazei alcaline este puternic crescută - de 2-3 ori. Aciditate crescută a sângelui |
| Urină | Testul Sulkovich pentru excreția de calciu este pozitiv. Prezența proteinelor, sângelui (mai mult de 2 - 3 eritrocite în câmpul vizual), leucocitelor. Azotul amino zahăr este de obicei normal | Testul lui Sulkovich este negativ. Proteine, fosfați, zahăr crescut, aminoacidurie |
| Oase | Zonele de calcificare sunt extinse, compactate | Osteoporoza oaselor tubulare, deficit de calciu în zonele de calcificare |
Specialisti care se ocupa de sindromul Fanconi si complicatiile acestuia: nefrolog, ortoped, hematolog, endocrinolog, urolog, oftalmolog, genetician.
Tratament
Terapia rațională, bine concepută poate reduce impactul asupra creierului, sistemului osos și organelor al pierderilor excesive de aminoacizi esențiali, minerale, glucoză și proteine excretate prin urină.
Prin urmare, tratamentul pentru sindrom vizează următoarele sarcini:
- Corecția maximă posibilă a deficitului de potasiu, bicarbonați, modificări ale echilibrului acido-bazic al sângelui pentru a reduce acidoza.
- Terapia diabetului cu fosfat (rahitism rezistent la D) cu accent pe inadmisibilitatea restricției de lichide.
- Tratamentul bolii de bază care provoacă dezvoltarea sindromului dobândit la adulți.
Medical
 Pentru a atenua pierderea de fosfor și calciu, se folosesc preparate speciale cu vitamina D, acestea sunt l,25 (OH) D3 și l (OH) D3.
Pentru a atenua pierderea de fosfor și calciu, se folosesc preparate speciale cu vitamina D, acestea sunt l,25 (OH) D3 și l (OH) D3.
Dozele inițiale de vitamina D3 pe zi 10-15 mii UI. Creșterea dozei se efectuează treptat, crescând-o la fiecare 12-14 zile (sub controlul testului Sulkovich și al conținutului de fosfor din sânge). În absența semnelor de intoxicație și a unei ușoare excreții de calciu în urină, este permisă creșterea dozei, ducând-o la 100-150 mii UI pe zi și continuarea terapiei la niveluri normale de fosfor și fosfatază alcalină în sânge. Când valorile lor se stabilizează, doza nu trebuie crescută în continuare.
Terapia cu vitamina D se desfășoară în mai multe cursuri pentru a preveni crizele de progresie a deformărilor rahitice ale oaselor.
Cea mai bună opțiune este utilizarea metaboliților activi D3 - Oxidevit (0,5 - 1,5 mcg pe zi), calciotriol (Rocaltrol).
Acestea includ preparate de calciu (gluconat de calciu pe zi până la 1,5 - 2 grame), fosfor (0,5 - 1 gram pe zi), fitină.
Dintre fosfații anorganici, se folosește amestecul Albright, luând-o 1 lingură mare de 4 până la 5 ori pe zi. Fosfații se folosesc sub formă de soluție și tablete în doze calculate la 10 mg pe kilogram de greutate corporală, de 4 ori pe zi (întotdeauna cu preparate de vitamina D pentru a evita hiperparatiroidismul).
Cu o deficiență pronunțată de potasiu, se folosesc Panangin, Asparkam.
Pentru orice programare, CBS sau starea acido-bazică a sângelui este monitorizată constant. În mod normal, sângele are o reacție ușor alcalină, iar pH-ul este în intervalul 7,35 - 7,45. Cu acidoză, când valoarea pH-ului scade sub 7,35, sângele capătă o aciditate crescută.
În aceste cazuri, este indicată o perfuzie intravenoasă dintr-o soluție de bicarbonat de sodiu 4% sau să bea o soluție (50-60 ml pe zi), care include acid citric - 2 grame, citrat de sodiu - 3 g, citrat de potasiu - 3,3 g, apă 100 ml. 1 ml din acest amestec alcalinizant conține 1 mmol de sodiu și potasiu. Aciditatea ridicată a sângelui este neutralizată și cu ajutorul bicarbonatului de sodiu (bicarbonat de sodiu).
Pe baza rezultatelor practice, unii experți recomandă Unithiol ca mijloc de creștere a activității enzimelor dependente de tiol.
Când se prescrie cistinoză: Dithiotrental la o rată de 25 mg pe kilogram de greutate a pacientului după 3 ore; Cisteamină în doză zilnică de 90 mg/kg.
Există rezultate pozitive din utilizarea penicilaminei, care scade concentrația de acid piruvic în sânge, reduce gradul de excreție a aminoacizilor și favorizează creșterea rezervelor alcaline din organism.
Anabolicele hormonale, inclusiv metiltestosteronul, au un efect bun asupra funcționării tubilor renali.
Tratamentul într-un spital este indicat pentru tulburări metabolice pronunțate, inclusiv hipo- și hiperglicemie, deformări ale scheletului.
Prevenirea
Prevenirea modernă a sindromului Fanconi congenital în prezența unei patologii similare în familie constă în consiliere genetică preliminară. Riscul de a dezvolta boala pentru așa-zișii frați, adică surori și frați, este de aproximativ 25%.
În forma secundară a sindromului, simptomele sale scad sau dispar complet cu tratamentul activ al bolii de bază sau al stării patologice dobândite.
Prognoza
Prognosticul bolii depinde de forma (primară, secundară) a sindromului, de severitatea manifestărilor și de începerea tratamentului.
De exemplu, simptomele unui sindrom dobândit dispar atunci când cauza provocatoare este eliminată.
În cazul patologiei congenitale, pe fondul absenței depozitelor de cistină în țesuturi, evoluția bolii nu reprezintă o amenințare gravă pentru viață, altfel, mai ales fără tratament adecvat, moartea pacientului este prezisă până la 10-20. ani de la creșterea insuficienței renale.
Cu toate acestea, chiar și cu modificări severe ale rinichilor: pielonefrită, nefrită tubulointerstițială, insuficiență renală, cu expunerea precoce a medicamentului la procesul patologic și terapie pe termen lung, se stabilește un anumit echilibru în homeostazie, în care prognosticul unei calități normale a vieții. de zeci de ani este bine.
În practica medicală, există anamneză când la copiii de 7-8 ani sindromul ereditar Fanconi a fost practic „oprit” odată cu debutul unei remisiuni pe termen lung, o îmbunătățire clară a stării copilului și chiar recuperarea.
În cazul încălcării funcției renale responsabilă de absorbția inversă a nutrienților, este detectat sindromul Fanconi. Al doilea nume al afecțiunii este sindromul de Toni-Debre-Fanconi. În patologie se observă glucoză, proteină c, apar tulburări metabolice. Boala este moștenită. Diagnosticul se bazează pe constatările clinice. Scopurile tratamentului sunt compensarea stărilor deficitare, eliminarea insuficienței renale.
La copii, cidru de Toni-Debre-Fanconi provoacă rahitism, întârziere fizică, slăbiciune a țesutului muscular.
Descriere și motive
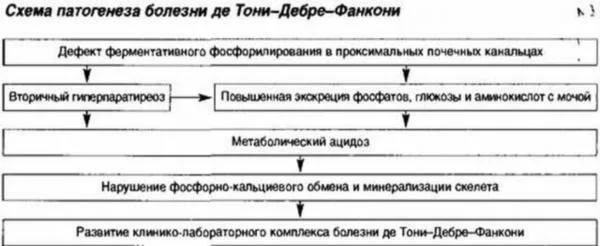
Boala De Toni-Debre-Fanconi se manifestă prin disfuncție tubulară severă, în urma căreia procesul de absorbție inversă a substanțelor și ionilor utili organismului este deranjat. În același timp, eliberarea de bicarbonați crește, sunt detectate defecțiuni metabolice generale. Particularitati:
- o scădere a conținutului de calciu cu fosfor în plasma sanguină;
- insuficiență în echilibrul acido-bazic, care se manifestă prin aciditate scăzută și bicarbonați în sânge;
- excreția normală a calciului sub formă de metabolit final în urină cu rate crescute de curățare naturală a organismului de fosfați;
- activitate patologic ridicată a fosfatazei alcaline;
- apariția zahărului în urină;
- o încălcare generală a producției și excreției produselor de sinteză a aminoacizilor;
- volume crescute de urină excretată cu densitate redusă.
 Boala Tony-Debre-Fanconi la copii.
Boala Tony-Debre-Fanconi la copii. Sindromul Fanconi se dezvoltă atât la copii, cât și la adulți. Există tipuri de patologie congenitale și dobândite. În funcție de severitatea simptomelor, precum și de tipul și severitatea tulburărilor metabolice, se disting 2 forme ale bolii:
- Primul tip se manifestă sub forma unei întârzieri pronunțate în dezvoltarea fizică cu o deformare clară a oaselor scheletului, un tablou clinic sever, fracturi frecvente pe fondul unei lipse accentuate de calciu din cauza absorbției slabe a substanței în intestinul.
- A doua variantă se manifestă sub forma unui grad moderat de întârziere a dezvoltării fizice, o manifestare ușoară a simptomelor cu distrugere osoasă slabă și un nivel suficient de calciu.
Cauzele bolii
Factorii provocatori sunt împărțiți în cei care provoacă forma dobândită a sindromului de Toni-Debre-Fanconi și cei care sunt responsabili de apariția patologiei congenitale. Următoarele condiții sunt recunoscute drept provocatori ai unei încălcări secundare:
- intoleranță la fructoză;
- lipsa enzimelor celulare;
- intoxicații cronice cu toxine;
- deficit sever de vitamina D.
Sindromul în sine este cauzat de o ereditate proastă sau de o nouă mutație a genei responsabile de procesele metabolice. Sindromul De Toni-Debre-Fanconi se dezvoltă adesea nu singur, ci împreună cu cistinoză, galactozemie, sindromul Wilson, tirozinemia primară și intoleranța la fructoză. Mai des, această formă este fixată la copii decât la adulți.
Factori provocatori
 Deficiența acută de vitamina D poate provoca boala.
Deficiența acută de vitamina D poate provoca boala. Sindromul Fanconi este mai probabil să apară la persoanele cu următoarele tulburări ereditare:
- insuficiență în procesele metabolice ale cistinei (aminoacizi din compoziția proteinelor);
- intoleranță la produsele lactate (chiar și laptele matern);
- defecte ale diferitelor enzime responsabile de sinteza și descompunerea glicogenului;
- eșec în schimbul de aminoacizi aromatici;
- intoleranță la fructoză;
- probleme cu metabolismul cuprului (boala Konovalov-Wilson);
- încălcarea metabolismului mielinei și disfuncția enzimei sulfatază;
- expunerea constantă la toxine din organism (droguri, otrăvuri, metale grele);
- eșec în procesele metabolice care implică proteine, ceea ce duce la depunerea unui complex specific - amiloid;
- deficit sever de vitamina D.
congenital și dobândit
Sindromul Fanconi este o boală severă asemănătoare rahitismului, care poate fi primară (congenitală) sau secundară (dobândită).
Patologia congenitală de Toni-Debre-Fanconi este ereditară, adică se transmite de la părinți la descendenți. Se dezvoltă adesea împreună cu alte tulburări genetice. Afecțiunea se caracterizează printr-o tulburare acută a proceselor metabolice, în urma căreia, împreună cu disfuncția tubilor renali, se dezvoltă orbirea, un sindrom hepatic mărit, un sindrom cu o scădere persistentă a concentrației și a activității tisulare a hormonilor tiroidieni. Mecanismul de moștenire al sindromului este determinat de tipul de patologie cu care se dezvoltă.
Sindromul de Toni-Debre-Fanconi dobândit este adesea provocat de diferite medicamente:
- medicamente pentru chimioterapie utilizate în tratamentul cancerului;
- medicamente antiretrovirale;
- tetraciclină învechită.
Provocatorii dezvoltării formelor secundare de patologie sunt transplantul complicat de rinichi, bolile oncologice ale sângelui, stările de deficit severe (în principal cu lipsă de vitamina D). Dacă o boală congenitală se dezvoltă în uter și este fixată în principal la copii, atunci forma secundară se poate dezvolta și la adulți.
Mecanism de dezvoltare și flux
 Reabsorbția substanțelor în rinichi este afectată.
Reabsorbția substanțelor în rinichi este afectată. Sindromul De Toni-Debre-Fanconi este cauzat de o mutație congenitală a genei responsabile de procesele metabolice, dar au fost observate cazuri de transformare proaspătă cauzată de alte patologii. Datorită malabsorbției multor substanțe și ioni utili, se dezvoltă stări severe de deficit:
- cu lipsa de aminoacizi, apare o distrofie severă, creșterea și dezvoltarea fizică încetinesc;
- la îndepărtarea fosforului și a bicarbonaților - un eșec al procesului de mineralizare cu debutul distrugerii țesutului osos;
- cu acumularea de zahăr în urină - o încălcare a reglementării metabolismului carbohidraților;
- când potasiul este excretat prin urină, atrofie musculară, o scădere a tensiunii arteriale la 80 mm Hg. Artă. si sub;
- cu tulburări metabolice pe scară largă - distrugerea țesutului renal (îngustarea lumenului cu aplatizarea epiteliului și scurtarea tubilor în care are loc reabsorbția).
Simptome la copii și adulți
Primele manifestări ale sindromului congenital de Toni-Debre-Fanconi apar în primul an de viață, mai rar - de la 1,5 ani. Copilul urinează adesea, există subfibralitate (până la 37-38 ° C), constipație, vărsături. Până la vârsta de 6 ani, dacă nu sunt tratați, copiii nu merg, iar până la vârsta de 12 ani, rinichii eșuează, ceea ce este plin de moarte. Pacienți - nesociabili, notori, timizi. Funcția mentală și cogitativă este normală. Consecințele sindromului sunt probleme cu Adunarea Națională și vederea, dezvoltarea imunodeficienței cronice, disfuncția organelor genito-urinale și a tractului gastrointestinal.
Simptomele la copii se dezvoltă în cea mai mare parte din cauza deficienței de fosfat (cum ar fi diabetul de fosfat) și se manifestă prin:
- instabilitate a mersului;
- mic de statura;
- mobilitate redusă;
- rotunjime a picioarelor;
- curbura oaselor scheletului (în special a coloanei vertebrale);
- dureri osoase, de ce copilul nu vrea să meargă;
- o creștere bruscă a volumului de urină eliberat;
- o scădere a greutății specifice pe fondul unei modificări calitative a compoziției urinei;
- atonia musculară;
- dureri osoase;
- boala hipertonică;
- insuficiență renală cronică (în absența tratamentului);
- osteoporoză, osteopenie pe fondul scăderii densității osoase.
Boala De Toni Debre Fanconi, numită în mod obișnuit sindromul Fanconi, este asociată cu afectarea funcției renale. Eșecurile care apar în corpul uman din cauza acestei boli duc la complicații. Acesta este urmat de un articol, ale cărui informații vor ajuta cititorul să înțeleagă caracteristicile bolii.
Ce este boala Fanconi
 Denumirea bolii provine de la numele doctorului elvețian Fanconi, care a fost primul care a studiat cauzele și simptomele bolii la începutul secolului trecut. Fanconi a examinat copii mici cu rahitism care prezentau modificări ale urinei. Medicul a colectat aceste manifestări împreună și le-a adus într-o singură patologie - sindromul Fanconi.
Denumirea bolii provine de la numele doctorului elvețian Fanconi, care a fost primul care a studiat cauzele și simptomele bolii la începutul secolului trecut. Fanconi a examinat copii mici cu rahitism care prezentau modificări ale urinei. Medicul a colectat aceste manifestări împreună și le-a adus într-o singură patologie - sindromul Fanconi.
Observând pacienții timp de doi ani, Tony a adăugat încă 2 factori patologici la factorii patologici existenți: o scădere a nivelului de fosfați din sânge sub norma stabilită și hiperaminoacidurie.
Patologia este moștenită. Urina pacientului în timpul analizei este umplută cu o cantitate în exces de zahăr, proteine și fosfați. De asemenea, metabolismul încetinește și se oprește absorbția produselor renale în sânge.
În lume, pentru fiecare 350 de mii de nou-născuți, există 1 sugar cu sindromul de mai sus. Este de remarcat faptul că adulții se îmbolnăvesc rar de acest defect.
Cauzele acestei boli
Astăzi, cauzele sindromului Fanconi pot fi diferite. Medicii sugerează că dezvoltarea bolii este asociată cu o mutație genică inexplicabilă, în timpul căreia funcțiile enzimelor se schimbă, conținutul de fosfor din sânge scade și activitatea aminoacizilor este perturbată. Unii sunt înclinați să concluzioneze că patologia este cauzată de disfuncția proteinelor tubulilor rinichilor, care se dezvoltă din cauza defectelor structurii mitocondriilor.
Alte cauze ale sindromului:
- intoxicații cu otrăvuri și toxine care apar ca urmare a ingerării de metale grele;
- deficit de vitamina D, provocând defecte la nivel genetic;
- eșecuri în asimilarea enzimelor celulare necesare organismului;
- depunerea în țesuturi de amiloid în timpul unei încălcări a metabolismului proteinelor;
- încălcarea procesului metabolic din organism, numită cistinoză;
- tirozinemia.
Multe studii arată, de asemenea, că boala este una dintre complicațiile rahitismului. Rahitismul apare din cauza lipsei de enzime celulare, în timp ce majoritatea substanțelor utile și necesare părăsesc corpul uman cu urină, transformând țesutul osos într-o masă înmuiată.
Cauzele exacte ale apariției sindromului Fanconi, în urma informațiilor de mai sus, nu au fost identificate, prin urmare, în medicină, patologia se numește diabet glucofosfamină.
Simptome care indică o patologie existentă
O manifestare pronunțată a patologiei este vizibilă în copilărie. Din momentul în care copilul se naște și până la un an, încep să apară primele semne:
- volum crescut al secretiilor urinare -;
- gâzâiala;
- apariția constipației;
- creșterea temperaturii corpului;
- sete constantă - polidipsie;
- slabiciune musculara;
- flatulență - gaze în intestine;
- durerea ascuțită în oase, senzația cu care copilul plânge în mod constant, poate fi confundată.
Boala De Toni Debre Fanconi la copii reduce abilitățile intelectuale. Conform datelor fizice, bebelușul rămâne în urma semenilor săi. Acest lucru este cauzat de lipsa de vitamine care sunt excretate din corpul copilului prin urină. Membrele inferioare devin similare datorită curburii literei „X”. Organele copilului scad în timpul dezvoltării atrofiei fibrelor musculare. Totul se termină cu faptul că la vârsta de 5 ani, copilul nu este capabil să facă un pas mic.
Tratamentul prematur duce la faptul că atunci când cresc la adolescenți, funcționalitatea rinichilor scade. Există, de asemenea, efecte secundare ale bolii: scăderea vederii, inima și vasele de sânge experimentează o încărcare crescută, afectarea sistemului nervos central.
Forme cunoscute ale bolii identificate
Boala este împărțită în 2 forme:
- Forma primară, când boala a fost moștenită de copil.
- Forma secundară, când o persoană s-a îmbolnăvit la vârsta adultă.
Cu patologia ereditară, defecte sunt găsite în cromozomul X. Mutația se transmite de tip recesiv și dominant. Specialiştilor le este greu să facă o prognoză exactă. Diagnosticul bolii este posibil numai în timpul alăptării. Într-un alt mod, forma primară a sindromului se numește „infantil.
Măsura în care cromozomul X a suferit mutații explică semne suplimentare. Puteți identifica cu exactitate boala prin 3 defecte principale:
- aminoacidurie.
Este important de știut! Dacă sunt detectate doar 2 componente ale tulburărilor de mai sus, diagnosticul se numește incomplet.
Metode de diagnosticare a bolii
Practic, sindromul Fanconi poate fi depistat folosind raze X și sânge. Radiografia arată următoarele:
- întârzierea creșterii la copii;
- fuziunea lentă a membrelor cu fracturi;
- rahiocampsis;
- deformare osoasa;
- emaciare în oasele tubulare;
- friabilitatea se formează în zonele de creștere ale membrelor osoase;
- scăderea densității osoase, timp în care acestea devin fragile.
Cu testele de sânge, patologia poate fi diagnosticată de următorii factori:
- exces de potasiu;
- niveluri crescute de hormon paratiroidian;
- concentrație scăzută de fosfor și calciu;
- creșterea acidității în organism;
- enzima fosfatază alcalină este crescută.
Analiza urinei indică sindromul Fanconi în cazul:
- exces de săruri de fosfat în urină;
- naturiurie;
- creșterea nivelului de aminoacizi în urină;
- glucoză crescută.
Pe o notă! În unele cazuri, examinarea radioizotopilor, nefrobiopsia și biopsia țesutului osos sunt utilizate pentru diagnostic.
Modalități de a trata această problemă
Medicii iau în serios tratamentul patologiei. Terapia terapeutică se desfășoară în complex: cu ajutorul medicamentelor și a intervențiilor chirurgicale. Adesea, la aceasta se adaugă dieta. În tratament, sarcina principală a medicului este de a corecta lipsa de fosfor din sânge și de a corecta deficiența de potasiu.
Dieta fara aminoacizi
Pacientul trebuie să respecte o dietă pentru a reduce intensitatea excreției aminoacizilor din organism împreună cu urina. Prin urmare, meniul va consta din următoarele produse:
- cartof;
- varză;
- lactate;
- fructe și sucuri din acestea;
- stafide;
- datele;
- prune uscate;
- caise uscate.
Medicul poate prescrie pacientului un aport suplimentar de medicamente vitaminice bogate în vitamina D și potasiu.
Intervenție chirurgicală
Pacientul stă sub cuțitul chirurgului în cazul unor defecte severe ale țesutului osos, din cauza cărora este imposibil să se facă mișcări simple. Operația este posibilă doar atunci când a venit faza de calm și observarea remisiunii persistă timp de 1,5 ani. Pentru atenuarea manifestărilor patologiei, pacientului i se recomandă masaj și băi cu ace de pin și sare de mare.
Tratament cu medicamente
Pentru a restabili conținutul normal de calciu și fosfor din sânge, pacientul, pe lângă dietă, ia medicamente prescrise de medic. Este la fel de important să luați un medicament al cărui element activ este vitamina D. La început, doza este de 10.000 UI pe zi.
În timp, doza va crește la 100.000 UI pe zi. Când testele arată conținutul normal de fosfor și calciu, aportul de vitamina D este anulat. În tratamentul formei secundare a bolii, se folosesc medicamente care conțin fitină și calciu.
Sindromul Fanconi, cunoscut și sub denumirea de boala Tony-Debre-Fanconi, este o afecțiune a rinichilor în care absorbția inversă a marii majorități a substanțelor este perturbată, care este însoțită de glucozurie (zahăr în urină), aminoacidurie (proteine), hiperfosfaturie ( fosfați) și se observă tulburări metabolice pronunțate în organism . Această afecțiune la copii duce la întârziere în dezvoltare, rahitism și slăbiciune musculară.

În sindromul Fanconi, reabsorbția substanțelor în rinichi este afectată.
Cauzele acestei afecțiuni sunt diverse și nu au fost încă precizate. De regulă, sindromul Fanconi este asociat cu intoleranța, deficiența unui număr de sisteme enzimatice celulare (fosfoenolpiruvat carboxikinaza, de exemplu), intoxicația cronică a corpului cu toxine și deficiența de vitamina D.
Din cauza diferenței de opinii, aceeași afecțiune poate fi numită și „sindrom Fanconi renal idiopatic” și „rahitism rezistent la D”. Uneori se întâlnește termenul „diabet glucofosfaminic” sau „nanism renal cu rahitism D-rezistent”.
Apare relativ rar, ceea ce, în parte, explică lipsa de informații despre acest sindrom. În medie, sindromul apare la unul din 350.000 de copii.
Varietăți de sindrom Fanconi

Pe fondul deficienței de fosfat, tibiele devin în formă de O
Alocați sindromul congenital (primar, idiopatic) și dobândit.
Sindromul Fanconi primar este considerat legat de X.
Poate fi dominant sau recesiv, de ex. moștenit într-o varietate de moduri, prezicerea prezenței descendenților este destul de dificilă.
Sindromul Fanconi determinat genetic poate fi complet și incomplet, adică. uneori apar doar 2 din cele 3 simptome clasice (glucozurie, aminoacidurie, hiperfosfaturie).

Sindromul Fanconi primar este o tulburare genetică
Secundar, de regulă, este o consecință a tirozinemiei de primul tip. Contribuie la apariția sindromului patologiilor ereditare ale rinichilor. Sindromul Fanconi se dezvoltă destul de des după organ (cu histocompatibilitate scăzută).
Boala Tony-Debre-Fanconi poate rezulta din otrăvirea cu mercur, uraniu, plumb sau cadmiu. Uneori se dezvoltă la lucrătorii din producția chimică, și anume la contactul cu toluenul, lizolul și acidul maleic.
Uneori, sindromul Fanconi se dezvoltă în timpul tratamentului cu medicamente cu platină, gentamicină și medicamente cu tetraciclină expirate.
Simptomele sindromului Fanconi
Galeria unor simptome
 Urinare frecventa
Urinare frecventa  Rachiocampsis
Rachiocampsis  Temperatură ridicată
Temperatură ridicată
La copii, de regulă, partea principală a simptomelor este asociată cu o deficiență de fosfat (similar cu diabetul fosfat). Există un mers tremurător („răță”), statură mică, inactivitate. Forma în formă de O a tibiei se formează treptat, alte oase ale scheletului (în special coloana vertebrală) sunt îndoite. Din cauza durerii de oase, copilul merge puțin. Riscul de fracturi din cauza țesutului osos este foarte mare.
Copiii cresc necomunicați și timizi - funcțiile cognitive (mentale) nu suferă, dar se formează complexe.

Constipația poate fi, de asemenea, asociată cu boala Tony-Debre-Fanconi.
Primele semne ale bolii în formă congenitală apar deja în primul an de viață (uneori de la 1,5 ani).
Copilul începe să urineze frecvent, apare (37-38 0C), se dezvoltă constipație, pot apărea vărsături.
De-a lungul timpului, părinții observă simptomele de mai sus ale deficitului de fosfor. Până la vârsta de 6 ani, copiii nu mai pot merge singuri. Odată cu dezvoltarea procesului până la vârsta de 12 ani, se formează, care este plin de un rezultat fatal.
Tulburările metabolice duc la patologii ale sistemului nervos, probleme de vedere, defecte în dezvoltarea organelor sistemului genito-urinar, boli intestinale și imunodeficiență cronică.
La adulți se manifestă sindromul secundar (urinat frecvent), slăbiciune generală, hipotensiune musculară și dureri osoase. Insuficiența renală se formează activ, se dezvoltă hipertensiunea arterială.
Cel mai rău dintre toate, atunci când sindromul Fanconi dobândit afectează femeile aflate în postmenopauză. Pe lângă scăderea naturală a densității osoase (osteopenie, osteoporoză), se adaugă fragilitatea osoasă din cauza deficienței minerale. Această situație se încheie cu fracturi de compresie ale corpurilor vertebrale, fracturi ale capului femural și invaliditate a pacientului.

Osteoporoza este un simptom auxiliar în diagnosticul sindromului Fanconi
Diagnosticul sindromului Fanconi
Sindromul este rar, un medic cu experiență îl poate suspecta din cauza radiografiei oaselor și a unor parametri biochimici avansați ai sângelui și urinei.

Apariția pintenilor este asociată cu o încălcare a procesului de formare a osului
Sindromul Fanconi este adesea combinat cu osteoporoza sistemică (osteoporoza este un simptom auxiliar în acest caz). Țesutul osos are o structură fibroasă grosieră, excrescențe osoase („pinteni”) sunt foarte des vizibile. Densitometria este utilizată pentru a determina densitatea osoasă.
Oasele sunt slab mineralizate, ceea ce se determină prin analiza biopsiei (probă de țesut).
Din partea rinichilor - în timpul studiului se determină distrofia tubilor renali (după tipul „gât de lebădă”), epiteliul este distrus și structurile renale sunt înlocuite cu țesut conjunctiv.
Singurul lucru care salvează pacienții este că stratul glomerular este ultimul implicat în proces, iar până la sfârșitul bolii, funcția rinichilor este cumva, dar dusă la îndeplinire.
Microscopia epiteliului glomerular arată un număr mare de mitocondrii în celule.
Tratamentul bolii
Sindromul Fanconi primar nu poate fi vindecat, deoarece vorbim de modificări structurale ale rinichilor, precum și de tulburări metabolice persistente, determinate genetic. Astfel de pacienți mențin constant nivelul de potasiu, tratează acidoza tubulară renală și alte defecte ale metabolismului apă-sare. Diabetul cu fosfat în acest caz este supus terapiei standard pentru această boală.
Pacienții trebuie să bea multe lichide pe parcursul zilei.
Galeria unor tratamente pentru sindromul Fanconi
 Picuratoare
Picuratoare  vitamine
vitamine  Băi de conifere
Băi de conifere  Masaj
Masaj
Sindromul Fanconi secundar în unele cazuri este vindecabil, mai ales cu eliminarea cu succes a bolii sau afecțiunii care provoacă această patologie renală.
Terapie medicamentoasă
Pentru a compensa tulburările metabolismului fosforului și calciului, sunt prescriși metaboliți ai vitaminelor din grupa D - 1 (OH) D3 sau 1,25 (OH) D3. Dozele de vitamine sunt titrate de la 10.000 UI la 100.000 UI pe zi. Doza este selectată sub monitorizarea constantă de laborator a nivelului de fosfor și calciu din sânge. Sunt prescrise preparate de calciu și fitină per os.
O astfel de terapie se efectuează periodic, în cursuri.
Dietoterapia pentru sindromul Fanconi
Principiul principal al dietei este limitarea excreției unui număr de substanțe din organism, inclusiv. aminoacizi care conțin sulf și fosfor. Dieta presupune limitarea sării și utilizarea pe scară largă a alimentelor alcalinizante.
Este necesar să se introducă în alimentație o cantitate mare de sucuri de fructe și lapte (cu toleranța sa normală)
 sucuri de fructe
sucuri de fructe  restricție de sare
restricție de sare  Lapte
Lapte  Fructe uscate
Fructe uscate
Dacă tulburările sistemului musculo-scheletic devin critice, atunci se recomandă corectarea chirurgicală.
Diagnosticul în timp util al tulburărilor metabolice în funcție de tipul de sindrom Fanconi la copii face posibilă evitarea dizabilității rapide a copilului, la adulți - pentru a reduce riscul de fracturi și lezarea rădăcinilor fibrelor nervoase, pentru a crește calitatea și viața. speranța. La primele semne ale bolii (poliurie, decolorarea urinei, dureri în oase și articulații), consultați un medic.
Sindromul Fanconi este o tulburare metabolică sistemică în care este detectată disfuncția rinichilor. Ei își pierd capacitatea de a reabsorbi și de a reveni în fluxul sanguin aminoacizii, glucoza, sărurile de calciu și fosfor excretate prin urină. Acesta este un defect organic congenital sau dobândit. Este extrem de rar, 1 caz apare la 350 de mii de nou-născuți. Cu toate acestea, această disfuncție este extrem de greu de afectat starea corpului.
Se mai numește și sindromul de Toni-Debre-Fanconi, diabetul fosfatic, sindromul Fanconi ereditar. Unii oameni de știință cred că această patologie apare atunci când rezervele de ATP (acid adenozin trifosforic) scad brusc în celule. Alții au prezentat versiunea conform căreia deformarea osoasă șubredă apare din cauza unei deficiențe de fosfor, a unei defecțiuni în echilibrul acido-bazic sau din cauza ambilor factori. Alții cred că disfuncția rinichilor nu se datorează unui defect biochimic, ci unui defect structural. Motivul exact nu a fost încă stabilit.
Cu această patologie, tubii proximali renali sunt afectați. Concentrația compușilor de calciu și potasiu în sânge rămâne în intervalul normal. Reacția urinei este alcalină sau neutră. Datorită pierderilor semnificative de hidrocarburi, se dezvoltă acidoză. Aceasta este o condiție periculoasă în care există o încălcare a echilibrului acido-bazic și „acidificarea” corpului. Acidoza este de obicei cauzată de orice boală difuză a rinichilor.
Patologia congenitală poate avea diferite grade de severitate. În prezența a 3 defecte biochimice, complet este diagnosticat, iar în cazul a 2 - disfuncție renală incompletă. Unii oameni de știință cred că sindromul Fanconi ereditar este o boală independentă asemănătoare rahitismului. Cu toate acestea, cel mai adesea însoțește alte patologii congenitale. Poate fi:
- cistinoză (o boală asociată cu cristalizarea aminoacidului cistinei în țesuturi);
- boala Wilson-Konovalov (o patologie cauzată de depozitele de cupru în rinichi, ficat, creier);
- galactozemie (conversia afectată a zahărului simplu în glucoză);
- sindromul Lowe (o patologie manifestată prin întârziere de creștere, dezvoltare mentală, cataractă și glaucom);
- tirozinemia (pasivitatea enzimei hepatice care descompune aminoacidul tirozina);
- intoleranță la fructoză.
Disfuncția dobândită, numită după Fanconi, poate apărea din cauza unor astfel de factori:
- medicamente care sunt toxice pentru rinichi (Aspirina, Gentamicină, Tetraciclină expirată, medicamente antivirale Cidofovir, Didanozină, medicamente antitumorale Streptozocin, Ifosfamidă etc.);
- intoxicații cu săruri de metale grele, alți compuși chimici agresivi;
- deficit acut de vitamina D;
- mielom multiplu (cancer de sânge);
- amiloidoză (tulburări ale metabolismului proteinelor);
- transplant de rinichi.
Simptomele bolii la copii
Cel mai pronunțat sindrom Fanconi congenital la copii. Simptomele sale se fac simțite deja în primul an de viață al bebelușilor. Acest:
- poliurie (urinat frecvent cu o cantitate mare de lichid excretat din organism);
- polidipsie (sete intensă patologic);
- temperatură ridicată;
- convulsii;
- vărsături frecvente fără motiv aparent;
- constipație prelungită;
- erupții cutanate;
- umflarea articulațiilor;
- mărirea rinichilor, a ganglionilor limfatici, a splinei;
- predispoziție la infecții.

Datorită excreției zilnice de vitamine, oligoelemente, glucoză din organism cu un lichid, copilul rămâne în urmă în dezvoltarea mentală și fizică. Oasele picioarelor lui sunt îndoite, devin flăcătoare, iar apoi mușchii se atrofiază complet. Adesea, copiii își pierd capacitatea de a merge independent. Până la vârsta de 12-13 ani, se dezvoltă insuficiență renală cronică. Uneori vederea se deteriorează. În plus, cu sindromul Fanconi, simptomele patologiei pot indica defecte congenitale combinate ale vaselor și inimii, ale organelor digestive și ale sistemului genito-urinar.
Simptomele bolii la adulți
Cel mai des remarcat:
- poliurie;
- hipostenurie (scăderea densității relative a urinei);
- dureri în oase;
- osteomalacie (înmuierea țesutului osos și pierderea rezistenței);
- slabiciune musculara;
- progresia accelerată a hipertensiunii arteriale;
- insuficiență renală cronică (dacă nu există tratament).
Poliuria în sindromul Fanconi la adulți nu este pronunțată, aceasta fiind semnificativ diferită de excesul de urină în diabetul insipid. Cel mai adesea, acest sindrom devine o manifestare a mielomului multiplu sau a bolii Waldenström (leziuni maligne ale sistemului hematopoietic). Pe lângă poliuria moderată, activitatea renală afectată se manifestă prin scăderea funcției de concentrare a acestora, apariția proteinelor în urină.
Dacă există sau nu sindromul Fanconi poate fi judecat după simptome caracteristice precum o concentrație mare de aminoacizi, glucoză și fosfați în urină. Excreția crescută a glucozei în urină în această patologie se datorează disfuncției renale. Cu toate acestea, concentrația de zahăr pe stomacul gol și nivelul de hiperglicemie la acești pacienți, de regulă, sunt în limitele normale.
Poliuria este întotdeauna însoțită de slăbiciune musculară, care se simte cel mai acut la nivelul membrelor. Apare din cauza deficitului de potasiu. În plus, aproape întotdeauna pacienții sunt afectați de dureri de oase.

Simptomele acidozei la adulți și la copii sunt aceleași. Acest:
- pierderea poftei de mâncare;
- vărsături fără cauză;
- constipație sau diaree;
- un miros acru care vine din piele sau din gură;
- reducerea presiunii;
- durere de cap;
- dispnee;
- insomnie;
- prosternare.
Cu toate acestea, medicii disting acidoza adultului de copilărie. Se crede că la sugari este întotdeauna o patologie congenitală. La adulți, poate fi atât o dezvoltare ulterioară a unei boli a copilăriei, cât și o complicație dobândită din cauza afectarii rinichilor. Simptomele acidozei sunt adesea combinate cu semne de nefrită interstițială și urolitiază. Pietrele la rinichi se formează din cauza pierderilor mari de calciu în urină. În cazurile severe, sunt detectate simptome de osteoporoză și osteomalacie.
Diagnosticul patologiei
Identificarea sindromului de Toni-Debre-Fanconi începe cu colectarea unei anamnezi, examinarea pacientului și teste de laborator. Semnele caracteristice ale patologiei în hemograma biochimică sunt nivelurile scăzute de calciu, fosfor, potasiu și sodiu, precum și glucoză, alanină, glicină, acid glutamic. Dar în urină - un nivel ridicat de fosfor și glucoză, există proteine și leucocite.
Cu o pierdere semnificativă de bicarbonați în urină și o acumulare excesivă de acizi în organism, este diagnosticată acidoza metabolică. Aproape toți pacienții au un exces al nivelului de acizi piruvic și lactic din sânge. De asemenea, este detectată o scădere a activității enzimelor din metabolismul energetic.
Cu diagnosticul instrumental, ecografia rinichilor și ureterelor este obligatorie. Nefrobiopsia (examinarea mostrelor miniaturale de rinichi) vă permite să vedeți deformarea tubilor proximali, care sunt alungiți ca gâtul unei lebede. În stadiile ulterioare ale bolii, este detectată atrofia glomerulilor renali.

Radiografia extremităților inferioare deformate ajută la detectarea degenerării țesutului osos. Fracturile ascunse ale cartilajului epifizar sunt adesea întâlnite la copii, din cauza cărora oasele nu mai cresc ulterior în lungime și devin asimetrice. În tibie sunt detectate neoplasme asemănătoare pintenilor.
În etapele ulterioare ale dezvoltării sindromului, osteoporoza este diagnosticată. Riscul de fracturi ale oaselor tubulare este mare. Stadiul osteoporozei este determinat prin densitometrie cu raze X. Mineralizarea scăzută a țesuturilor osoase este detectată în studiul probelor acestora, obținute tot prin biopsie.
Principalele criterii de diagnostic:
- Un deficit semnificativ în greutatea și înălțimea copilului.
- Slăbiciunea funcțiilor statico-motorii.
- Deformări asemănătoare rahitismului ale scheletului (degradarea structurii țesutului osos este confirmată de raze X).
- tulburări electrolitice.
Cu sindromul Fanconi la adulți, medicii trebuie să efectueze un diagnostic diferențial pentru a exclude patologii similare cu acesta. Acest:
- boli ereditare (cistinoză, galactozemie, boala Wilson-Konovalov, tirozinemia etc.);
- pielonefrită cronică;
- Diabet;
- hiperparatiroidism secundar;
- mielom multiplu;
- intoxicații cu medicamente, substanțe chimice agresive;
- arsuri extinse.
Tratamentul patologiei
În mod ideal, atunci când sindromul Fanconi este tratat de un genetician, un hematolog, dar nu orice unitate medicală are astfel de specialiști. Cel mai adesea, această patologie este tratată de un nefrolog sau urolog. Dacă se observă semne de hiperparatiroidism (hipersecreție a glandelor paratiroide), este necesară consultarea unui endocrinolog. Dacă vederea se deteriorează, este necesar să fie examinat de un oftalmolog.

Tacticile de tratament includ:
- Suplimentarea deficitului de electroliți.
- Eliminarea încălcărilor echilibrului acido-bazic.
- Terapia pentru insuficienta renala.
- Îndepărtarea simptomelor dureroase.
Pentru tratamentul medical, se folosesc următoarele:
- Calcitriol, Oxidevit și alte preparate cu vitamina D;
- gluconat de calciu;
- Fitin, glicerofosfat de calciu, fosfat de aluminiu;
- solutii alcalinizante Bizitra, Polizitra;
- Indometacin, Metiltestosteron, Hipotiazidă (cu afectare severă a tubilor proximali);
- Panangin, Asparkam;
- Cistina, Mercaptamina;
- antibiotice;
- corticosteroizi etc.
Deoarece sindromul Fanconi este cronic, tratamentul se efectuează pe o perioadă lungă de timp, în cure mari după pauze obligatorii. Adesea, este posibil să aduceți metabolismul mai aproape de normă, să atenuați severitatea manifestării bolii și să preveniți complicațiile periculoase. Dar este extrem de dificil să scapi complet de sindromul de Toni-Debre-Fanconi, de multe ori dă recidive.
Terapia sindromului are ca scop eliminarea acidozei, completarea rezervelor de potasiu, bicarbonat de calciu, fosfați și alți electroliți. Cel mai important rol în acest sens îl joacă o dietă terapeutică. Trebuie să beți multă apă și să reduceți cantitatea de sare. Mănâncă mese mici, dar des. Carbohidrații trebuie dozați strict, astfel încât să nu existe exces de glucoză în sânge.
Articole similare
-
Când un soț este împotriva unui copil, cum să rămâi însărcinată fără știrea lui?
Uneori poți rămâne însărcinată din neglijență. Pentru a preveni acest lucru, este important să știți cum puteți concepe un copil accidental și ce mijloace puteți utiliza pentru a evita o sarcină nedorită. De asemenea, în acest articol puteți găsi informații despre...
-
Ce pietre și amulete sunt potrivite pentru Taur în funcție de horoscop și data nașterii Talisman de elefant pentru Taur
Aprilie-mai Taurul (21 aprilie - 20 mai) sunt măsurați, nu mofturoși și colosal de productivi! Încăpăţânarea lor de invidiat îi poate aduce pe alţii la mâner, dar ştiu exact ce fac şi de ce au nevoie. Printre aspectele pozitive...
-
Restricții privind accesul la date în roluri 1c
Toate setările pentru drepturile utilizatorului pe care le vom face în cadrul acestui articol se află în secțiunea 1C 8.3 „Administrare” - „Setări pentru utilizatori și drepturi”. Acest algoritm este similar în majoritatea configurațiilor pe...
-
1c pornește un client subțire în loc de unul gros
Platforme: 1C: Enterprise 8.3, 1C: Enterprise 8.2, 1C: Enterprise 8.1 Configurații: Toate configurațiile2012-11-16 21362 Sunt lansate prin specificarea specială...
-
Dovezi despre modalități cunoscute de a fura energie electrică Cum să afli cine fură electricitate
Creșterea tarifelor la energie este una dintre trăsăturile izbitoare ale adâncirii crizei economice. În acest context, furtul de energie electrică și problemele asociate cu detectarea acesteia sunt de o importanță capitală. Modalități de a detecta furtul...
-
Caracteristici de montare prize și întrerupătoare pe diferite suprafețe
Salutări tuturor cititorilor blogului nostru.Astăzi, dragi cititori, vreau să subliniez subiectul cum se instalează prize. Această procedură este foarte des solicitată atunci când înlocuiți o priză veche cu una nouă în cazul unei defecțiuni, când ...