Objawy zespołu Gauchera. Choroba Gauchera: przyczyny, objawy, leczenie. Leczenie tej patologii
Reakcja Gauchera jest chorobą genetyczną polegającą na niedoborze enzymu biorącego udział w procesach metabolicznych na poziomie komórkowym. W miarę rozwoju choroby w tkankach i narządach gromadzą się komórki patologiczne.
Krótka wycieczka do historii
Reakcja Gauchera – co to jest? Chorobę po raz pierwszy zidentyfikowano w 1882 roku, kiedy francuski lekarz Charles Philippe Gaucher opisał jej objawy u pacjenta z powiększoną śledzioną.
W 1924 roku lekarzom udało się zsyntetyzować substancję tłuszczową zawierającą komórki patologiczne, tworząc w ten sposób pogląd na główny czynnik sprawczy choroby.
W 1965 roku specjaliści z Amerykańskiego Narodowego Instytutu Zdrowia udowodnili, że powstawanie niezdrowych komórek następuje na skutek dziedzicznego niedoboru w organizmie enzymu glukocerebrozydazy. Wyniki pomyślnej diagnozy posłużyły jako podstawa do opracowania jednej metody terapii poprzez substytucję enzymatyczną. Zastosowanie nowego podejścia do leczenia nie wyeliminowało całkowicie choroby, ale dało szansę na znaczne zmniejszenie jej objawów.
Reakcja Gauchera: socjologia. Co to jest?

Badania socjologiczne pokazują, że w populacji liczącej 100 000 osób mniej niż 1% osób ma patologiczną dziedziczność powodującą rozwój choroby. Wskazana częstotliwość nieznacznie wzrasta wśród ludności żydowskiej – imigrantów z regionu Europy Wschodniej. Dlatego ogólnie przyjmuje się, że reakcja Gauchera jest chorobą dziedziczną reprezentowanej narodowości. W rzeczywistości objawy patologiczne mają tę samą częstotliwość występowania, co hemofilia i inne choroby, w których wpływają lizosomy komórkowe.
Typy

Eksperci wyróżniają kilka rodzajów choroby:
- Bez neuropatii jest to najczęstszy rodzaj choroby, który u większości ludzi przebiega bezobjawowo. Przy poważnym osłabieniu organizmu objawia się przyspieszonym wzrostem patologicznej tkanki. Jednocześnie niezdrowe komórki nie wpływają na układ nerwowy i mózg.
- Z ostrymi objawami neuropatycznymi - niezwykle rzadki rodzaj choroby. Charakteryzuje się występowaniem wyraźnych patologii neurologicznych już w pierwszych latach życia. Statystyki pokazują, że bez odpowiedniej, terminowej diagnozy i opracowania odpowiednich metod leczenia śmierć następuje przed ukończeniem drugiego roku życia.
- W przypadku przewlekłej neuropatii - choroba objawia się powolnym postępem objawów patologicznych, obecnością umiarkowanych objawów neurologicznych. W późniejszych stadiach rozwoju wzrost niezdrowych komórek prowadzi do powiększenia narządów wewnętrznych, uszkodzenia układu oddechowego i rozwoju demencji. Jak poważna jest przewlekła reakcja Gauchera? Socjologia pokazuje, że większość pacjentów dożywa wieku dorosłego.
Diagnostyka

Wykrycie patologii wymaga kompleksowego zbadania organizmu. W szczególności postawienie prawidłowej diagnozy wymaga wizyty u neurologa, pediatry lub specjalisty z zakresu genetyki. Obecnie stosuje się kilka skutecznych metod, których wyniki mogą wskazywać na rozwój choroby:
- Badanie krwi jest najdokładniejszą metodą diagnostyczną, pozwalającą określić ilość enzymu glukocerebrozydazy w mikroskopijnych leukocytach i fibroblastach.
- Analiza DNA – pozwala na identyfikację genetycznych mutacji komórkowych. Postawienie diagnozy tą metodą możliwe jest z dokładnością do 90% już na etapie rozwoju płodu w łonie matki.
- Badanie szpiku kostnego ma na celu wykrycie zmian patologicznych w strukturze tkanek charakterystycznych dla choroby. Zastosowanie podejścia diagnostycznego umożliwia potwierdzenie choroby, ale nie pozwala na określenie lokalizacji komórek niosących zmutowane geny.
Objawy i objawy choroby
Jak wspomniano wcześniej, we wczesnych stadiach reakcja Gauchera przebiega bezobjawowo. W tym przypadku brak enzymu glukocerebrozydazy nie wpływa na funkcjonowanie narządów wewnętrznych i nie wpływa na układ nerwowy.
W zaawansowanej postaci choroba ma ostry, postępujący charakter. Na tym etapie pacjent odczuwa ogólne złe samopoczucie i okresowo cierpi na bóle brzucha, ponieważ wzrost komórek patologicznych wpływa przede wszystkim na śledzionę i wątrobę. Narządy te zwiększają swoją objętość, a przy braku właściwej diagnostyki i leczenia często ulegają pęknięciom tkanek.
W ten sposób objawia się reakcja Gauchera. W socjologii taki termin nie istnieje, choć postać Willa Smitha z filmu Focus twierdziła coś przeciwnego.
Początek objawów choroby w dzieciństwie często prowadzi do rozwoju patologii kości. Kości dziecka rozwijają się powoli, występują opóźnienia we wzroście i tworzeniu szkieletu.
Reakcja Gauchera: psychologia

Jednym z głównych problemów, z jakim borykają się osoby podatne na choroby genetyczne, jest stan ogólnego złego samopoczucia. To z kolei powoduje całą masę problemów psychologicznych. Osoby cierpiące na tę chorobę w ostrej postaci odczuwają zmęczenie nawet po całonocnym odpoczynku.
U dzieci reakcja Gauchera powoduje brak wytrzymałości i koncentracji. Z tego powodu trudno im komunikować się z rówieśnikami, bawić się z przyjaciółmi, uprawiać sport, skupiać się na zadaniach naukowych i uczestniczyć w życiu społecznym.
Zmiana wyglądu może prowadzić do pogorszenia stanu psychicznego pacjenta. Dziecko może być wyśmiewane z powodu niewystarczającego wzrostu, pełności lub słabo rozwiniętej masy mięśniowej. Osoby, które były narażone na ostre objawy choroby, często cierpią z powodu rozbieżności między danymi zewnętrznymi a pożądanym obrazem siebie. Konsultacja z psychologiem może pomóc w wyeliminowaniu powyższych problemów.
Możliwe konsekwencje
Nieleczone konsekwencje choroby obejmują zwykle:
- obfite krwotoki w narządach trawiennych;
- uszkodzenie tkanki wątroby i śledziony;
- skurcze krtani, aż do całkowitego ustania oddechu;
- rozwój niewydolności oddechowej, częste zapalenie płuc;
- występowanie procesów destrukcyjnych w tkance kostnej, złamania;
- zakażenie szpiku kostnego.
Leczenie

Jeszcze nie tak dawno terapia polegała jedynie na eliminowaniu głównych objawów choroby. Od lat 90-tych ubiegłego wieku jako główną metodę leczenia stosuje się enzymatyczną terapię zastępczą, która polega na wprowadzeniu do organizmu zmodyfikowanego pierwiastka glukocerebrozydazy. Sztuczny enzym kopiuje funkcje naturalnego składnika struktury krwi i kompensuje jego niedobór w organizmie. Zastrzyki zawierające substancję mogą wyeliminować negatywne objawy choroby, a w niektórych przypadkach zatrzymać tworzenie się komórek patologicznych jako całości.
Aby złagodzić ogólny stan, chorym przepisuje się leki przeciwbólowe. W miarę postępu choroby uciekają się do usunięcia części wątroby lub śledziony. W niektórych przypadkach przeszczep szpiku kostnego może rozwiązać problem.
Wreszcie
Reakcja Gauchera – co to jest? Jak widać, patologia jest zaburzeniem o dość niejasnych objawach. Na powodzenie leczenia bezpośrednio wpływa wykrycie patologii we wczesnych stadiach i wczesne rozpoczęcie terapii zastępczej. Opóźniona reakcja na problem prowadzi do rozwoju poważnych powikłań, a brak odpowiedniego leczenia najczęściej kończy się śmiercią.
to choroba genetyczna charakteryzująca się zaburzeniami metabolizmu lipidów, niedoborem enzymów lizosomalnych i nagromadzeniem glikolipidów w strukturach komórkowych. Objawy zależą od rodzaju patologii. Typowymi objawami są powiększenie wątroby, śledziony i zmniejszona krzepliwość krwi. W typie I stwierdza się zaburzenia układu kostnego: osteoporozę, częste złamania, infekcje kości. W typach II i III dominują objawy neurologiczne: drgawki, porażenie, zez, upośledzenie umysłowe. Diagnoza opiera się na analizie biochemicznej niedoboru enzymu. Leczenie obejmuje wymianę enzymów, redukcję substratu i leczenie objawowe.
ICD-10
E75.2 Inne sfingolipidozy

Informacje ogólne
Choroba wzięła swoją nazwę od nazwiska francuskiego lekarza Philippe’a Gauchera. W 1882 roku opisał objawy i cechy patologiczne budowy śledziony pacjenta zmarłego na sepsę. Kilkadziesiąt lat później, w podobnym przypadku klinicznym, Gaucher stwierdził akumulację glukocerebrozydu w śledzionie i niedobór enzymu glukocerebrozydazy. Choroba Gauchera (sfingolipidoza, lipidoza glukozyloceramidowa) należy do grupy lizosomalnych chorób spichrzeniowych - patologii dziedzicznych, w których zmieniają się funkcje organelli komórkowych lizosomów. Częstość występowania tej choroby waha się od 1:40 tys. do 1:70 tys. Największa częstość występowania występuje w społecznościach, w których dopuszczalne są małżeństwa między bliskimi krewnymi, np. wśród Żydów aszkenazyjskich. Noszenie mutacji genu stwierdza się u około 1 osoby na 400.
Powoduje
Sfingolipidoza glukozyloceramidowa jest najczęstszą postacią dziedzicznych enzymopatii. Za przyczynę jej rozwoju uważa się defekt genu GBA, kodującego enzym lizosomalny beta-glukozydazę (glukocerebrozydazę), odpowiedzialny za rozkład lipidów. Dziedziczenie choroby następuje w sposób autosomalny recesywny, do powstania fermentopatii konieczna jest obecność pary zmienionych genów: jednego od matki, drugiego od ojca. W małżeństwie, w którym oboje rodzice są nosicielami mutacji, prawdopodobieństwo urodzenia chorego dziecka wynosi 25%. Ryzyko przekazania jednego wadliwego genu, czyli ryzyko nosicielstwa bez rozwoju choroby w takich rodzinach, wynosi 50%. Jeśli w genotypie występują dwa zmutowane allele, funkcja glukocerebrozydazy jest zmniejszona o 15-30% normalnego poziomu.
Patogeneza
Patogenetyczną podstawą choroby jest zmniejszenie aktywności katalitycznej beta-glukozydazy. W rezultacie rozkład glikosfingolipidów (złożonych związków lipidów i węglowodanów) na glukozę i ceramidy zostaje zakłócony. Nieprawidłowo postępująca akumulacja makrocząsteczek następuje w komórkach charakteryzujących się zwiększoną szybkością ich odnowy – w makrofagach. Niehydrolizowane lipidy gromadzą się w lizosomach i powstają specjalne komórki magazynujące - komórki Gauchera. Pierwotna niewydolność metaboliczna powoduje wtórne zaburzenia procesów biochemicznych i funkcji komórkowych. Z powodu patologii metabolizmu tłuszczów rozwija się zespół aktywacji makrofagów. Pobudzona zostaje monocytopoeza, wzrasta zawartość makrofagów w wątrobie, śledzionie i szpiku kostnym. Powoduje to powiększenie śledziony, powiększenie wątroby i naciek szpiku kostnego. Zaburzenie funkcji regulacyjnej makrofagów jest czynnikiem wywołującym cytopenię, uszkodzenie kości i stawów.
Objawy choroby Gauchera
W zależności od wieku zachorowania i charakterystyki obrazu klinicznego wyróżnia się trzy typy choroby. Pierwszy typ jest najczęstszy i ma przebieg przewlekły. Objawy często pojawiają się w wieku 30-40 lat, rzadziej choroba objawia się w dzieciństwie. Zwiększenie wielkości wątroby i śledziony rozpoczyna się natychmiast po urodzeniu, ale klinicznie objawia się później. Pierwszymi oznakami patologii są niedokrwistość i zwiększone krwawienie. Hamowaniu układu krwiotwórczego towarzyszy spadek poziomu hemoglobiny i płytek krwi. Zmiany w układzie mięśniowo-szkieletowym objawiają się bólem kości i stawów, częstymi złamaniami i deformacjami (z reguły zmiany kości udowej). U dorosłych przebarwienia są widoczne na twarzy i nogach: skóra ciemnieje, przybierając odcień od żółtawego do żółtobrązowego. Mogą pojawić się płaskie czerwone plamy, zwykle zlokalizowane w okolicy oczu. Wzrost pacjentów jest poniżej średniej.
Drugi typ choroby (ostra wczesnodziecięca lub ostra neuropatyczna) jest bardzo rzadka, rozwija się od urodzenia do półtora roku życia, najczęściej objawy pojawiają się w pierwszych trzech miesiącach życia. Charakteryzuje się szybkim przebiegiem i słabą odpowiedzią na leczenie. Na pierwszy plan wysuwają się zaburzenia neurologiczne spowodowane nagromadzeniem komórek Gauchera w ośrodkowym układzie nerwowym. Dzieci słabo płaczą i słabo ssą. Odruch połykania jest osłabiony, często obserwuje się zaburzenia w cyklu oddechowym. Zauważalne jest opóźnienie w rozwoju psychicznym i fizycznym. W początkowej fazie choroby napięcie mięśniowe jest zmniejszone, po 9-12 miesiącach od wystąpienia pojawia się hipertoniczność, szczególnie w mięśniach szyi i kończyn. Rozwijają się drgawki, zez i porażenie spastyczne. Wątroba i śledziona są powiększone. Dzieci często cierpią na ciężkie zapalenie płuc.
Trzeci typ to neuropatia młodzieńcza lub podostra. Pierwsze objawy - powiększenie śledziony i wątroby - pojawiają się po 2-3 latach. Pełne objawy rozwijają się w wieku od 6 do 15 lat. Do klinicznych objawów uszkodzenia ośrodkowego układu nerwowego zalicza się wzmożone napięcie mięśni, porażenie spastyczne, zez, mimowolne skurcze, drgawki, trudności w oddychaniu z trudnościami w wdychaniu i problemy z połykaniem. Występują zaburzenia rozwoju psychicznego: obniżone funkcje intelektualne, niedojrzałość mowy i pisania, chwiejność emocjonalna, psychoza. Dzieci są opóźnione w rozwoju seksualnym. Przebieg choroby jest stale postępujący.
Komplikacje
Najpoważniejsze powikłania występują w drugim i trzecim typie choroby. Uszkodzenie rdzenia kręgowego i mózgu prowadzi do zakłócenia cyklu oddechowego, rozwijają się nagłe zatrzymania oddechu, wzrasta ryzyko skurczu krtani i śmierci w wyniku uduszenia. Obniżony poziom płytek krwi może powodować rozległe krwawienie wewnętrzne. U pacjentów z patologią typu 1 częstym powikłaniem jest zniszczenie kości, zwiększona łamliwość i zmiany zakaźne. Możliwość poruszania się jest ograniczona, pacjenci nie mogą poruszać się samodzielnie i wymagają opieki zewnętrznej.
Diagnostyka
Wywiad i badanie fizykalne przeprowadza endokrynolog i neurolog, zaleca się dodatkowe konsultacje z genetykiem, hematologiem, okulistą, pediatrą i psychiatrą. Dane anamnestyczne obejmują obecność choroby Gauchera u krewnych. Podczas badania ujawniają się typowe objawy: niski wzrost, patologie kości, objawy neurologiczne (zez, ataksja, porażenie), zespół krwotoczny, przebarwienia skóry. Czasami podejrzenie choroby pojawia się po przypadkowym wykryciu powiększonej śledziony na obrazach USG lub supresji układu krwiotwórczego zgodnie z ogólnym badaniem krwi. Aby potwierdzić diagnozę i wykluczyć inne dziedziczne patologie metaboliczne, zapalenie kości i szpiku, gruźlicę kości, wirusowe zapalenie wątroby i onkologiczne zmiany krwi, przeprowadza się szczegółową diagnostykę:
- Kliniczne, biochemiczne badanie krwi. Większość pacjentów cierpi na trombocytopenię, leukopenię i niedokrwistość, która u dzieci jest zwykle spowodowana niedoborem żelaza. Wyniki analizy biochemicznej wskazują na obniżoną aktywność glukocerebrozydazy.
- Analiza enzymatyczna komórek. W chorobie Gauchera niewystarczającą aktywność glukozydazy stwierdza się w suszonych próbkach krwi i fibroblastach skóry. Stopień niedoboru enzymu nie koreluje bezpośrednio z nasileniem objawów. Dodatkowym markerem biochemicznym jest chitotriozydaza. Enzym ten jest syntetyzowany przez aktywowane makrofagi, jego aktywność wzrasta zazwyczaj 6-10 razy.
- Badanie morfologiczne szpiku kostnego. Potwierdzono obecność struktur specyficznych dla tej choroby – komórek Gauchera. Wynik pozwala wykluczyć hemoblastozę i chorobę limfoproliferacyjną.
- Badanie struktury tkanki kostnej. W celu oceny stopnia uszkodzenia układu kostno-stawowego wykonuje się densytometrię, radiografię i/lub rezonans magnetyczny kości szkieletowych. Możliwa jest rozproszona osteoporoza; można uwidocznić kolby Erlenmeyera, ogniska osteolizy, osteosklerozę i martwicę kości. We wczesnych stadiach choroby obserwuje się osteopenię i naciek szpiku kostnego.
- Badanie obrazowe śledziony i wątroby. Wykonuje się USG i MRI narządów wewnętrznych. Na podstawie wyników określa się obecność lub brak zmian ogniskowych oraz mierzy się objętość powiększonego narządu. Wstępne wskaźniki pozwalają następnie monitorować skuteczność terapii.
- Molekularne badania genetyczne. Diagnostyka DNA jest procedurą opcjonalną. Potwierdzenie mutacji w genie GBA może być konieczne w przypadku, gdy badania biochemiczne są niejednoznaczne, a także w ramach badań prenatalnych i przedimplantacyjnych.
Leczenie choroby Gauchera
Specjalistyczna opieka nad pacjentami z I i III typem choroby ma na celu eliminację objawów i kompensację pierwotnego defektu genetycznego - zwiększenie ilości brakującego enzymu, zwiększenie katabolizmu glikosfingolipidów. W przypadku patologii typu 2 środki terapeutyczne nie są wystarczająco skuteczne, wysiłki lekarzy ograniczają się do łagodzenia objawów klinicznych - bólu, skurczów, zaburzeń oddechowych. Ogólny schemat obejmuje następujące obszary:
- Enzymatyczna terapia zastępcza. Główną metodą leczenia jest dożywotnia enzymatyczna terapia zastępcza (ERT) z wykorzystaniem rekombinowanej glukocerebrozydazy. Skuteczność jest dość wysoka - objawy ustępują całkowicie, poprawia się jakość życia pacjentów. ERT jest odpowiednia dla trzeciego i pierwszego typu choroby. Leki podaje się dożylnie. Częste wlewy czasami powodują choroby zapalne żył (zapalenie żył).
- Terapia redukująca substrat. Kierunek ten jest nowy w leczeniu choroby Gauchera i jest stosunkowo rozpowszechniony w USA i krajach Europy. Ma na celu zmniejszenie tempa produkcji substratu glikosfingolipidów i przyspieszenie katabolizmu akumulujących się makrocząsteczek. Stosowane leki są swoistymi inhibitorami syntazy glukozyloceramidowej. Metoda jest wskazana w przypadku choroby typu 1 z objawami łagodnymi do umiarkowanych.
- Terapia objawowa. W przypadku osteoporozy zalecana jest kompleksowa terapia obejmująca przyjmowanie leków zawierających wapń, witaminę D oraz przestrzeganie diety wzbogaconej w wapń. Środki te mogą spowolnić utratę masy kostnej, zwiększyć wytrzymałość kości i zapobiec złamaniom. W przypadku powikłań kostnych stosuje się leki przeciwbólowe (NLPZ) i terapię przeciwbakteryjną. Objawy zaburzeń neurologicznych można złagodzić za pomocą leków przeciwpadaczkowych, nootropowych i zwiotczających mięśnie.
Rokowanie i zapobieganie
Pozytywny wynik jest najprawdopodobniej u pacjentów z typem 1 choroby - zintegrowane podejście terapeutyczne pozwala na normalizację funkcjonalności glukocerebrozydazy, zapobieganie rozwojowi powikłań i uniknięcie niepełnosprawności. W typie 3 rokowanie zależy od charakteru choroby i indywidualnej reakcji organizmu na leczenie. Typ 2 ma niezwykle poważne objawy i kończy się śmiercią pacjenta. Profilaktykę prowadzi się na etapie planowania ciąży oraz w jej początkowej fazie. Rodzinom, których bliscy krewni cierpią na tę patologię, zaleca się medyczną poradnię genetyczną. Jeżeli istnieje duże ryzyko przeniesienia mutacji na nienarodzone dziecko w pierwszym trymestrze ciąży, wykonuje się badanie poziomu enzymów w płynie owodniowym i wydanie
RCHR (Republikańskie Centrum Rozwoju Zdrowia Ministerstwa Zdrowia Republiki Kazachstanu)
Wersja: Protokoły kliniczne Ministerstwa Zdrowia Republiki Kazachstanu – 2016
Inne sfingolipidozy (E75.2)
Choroby sieroce
informacje ogólne
Krótki opis
Zatwierdzony
Wspólna Komisja ds. Jakości Opieki Zdrowotnej
Ministerstwo Zdrowia i Rozwoju Społecznego Republiki Kazachstanu
od 29 września 2016 r
Protokół nr 11
Choroba Gauchera (GD)- lizosomalna choroba spichrzeniowa, choroba wieloukładowa, której podłożem jest niedobór enzymu glukocerebrozydazy, prowadzący do postępującego rozrostu narządów miąższowych, stopniowej infiltracji szpiku kostnego przez makrofagi obciążone lipidami, głębokich zaburzeń hematopoezy, a w niewielkiej część pacjentów z uszkodzeniem centralnego układu nerwowego.
Korelacja kodów ICD-10 i ICD-9:
Data opracowania protokołu: 2016
Użytkownicyprotokół: Lekarze pierwszego kontaktu, pediatrzy, onkohematolodzy.
Skala poziomu dowodu:
| A | Wysokiej jakości metaanaliza, systematyczny przegląd badań RCT lub duże badania RCT z bardzo niskim prawdopodobieństwem (++) błędu systematycznego, których wyniki można uogólnić na odpowiednią populację. |
| B | Wysokiej jakości (++) systematyczny przegląd badań kohortowych lub kliniczno-kontrolnych lub wysokiej jakości (++) badań kohortowych lub kliniczno-kontrolnych o bardzo niskim ryzyku błędu systematycznego lub RCT o niskim (+) ryzyku błędu systematycznego, wyniki które można uogólnić na odpowiednią populację. |
| C | Badanie kohortowe lub kliniczno-kontrolne lub badanie kontrolowane bez randomizacji z niskim ryzykiem błędu systematycznego (+). których wyniki można uogólnić na odpowiednią populację lub RCT z bardzo niskim lub niskim ryzykiem błędu systematycznego (++ lub +), których wyników nie można bezpośrednio uogólnić na odpowiednią populację. |
| D | Seria przypadków lub niekontrolowane badanie lub opinia eksperta. |
Klasyfikacja
Klasyfikacja
Ze względu na obecność i charakterystykę przebiegu klinicznego oraz zajęcia ośrodkowego układu nerwowego (OUN) dzieli się je na trzy typy choroby Gauchera:
· Nieneuropatyczny (typ I).
− Ityp -BbolesnyIGauchera jest najczęstszą postacią choroby, która nie wpływa na ośrodkowy układ nerwowy (stąd ten typ nazywany jest również chorobą nieneuropatyczną).
Objawy są niezwykle zróżnicowane – od postaci bezobjawowych po poważne uszkodzenia narządów i kości. Pomiędzy tymi biegunowymi grupami klinicznymi znajdują się pacjenci z umiarkowanym powiększeniem śledziony i prawie prawidłowym składem krwi, ze zmianami kostnymi lub bez. Chociaż ten typ choroby jest czasami nazywany chorobą Gauchera u dorosłych, może dotknąć osoby w każdym wieku. Im wcześniej pojawią się objawy kliniczne, tym poważniejsza jest choroba.
· Neuropatyczny (typ II iIII).
− II typ- ostra neuropatia. Choroba Gauchera typu 2 jest bardzo rzadką, szybko postępującą chorobą charakteryzującą się poważnym uszkodzeniem mózgu, a także prawie wszystkich narządów i układów.
Choroba typu 2, wcześniej nazywana chorobą Gauchera noworodków, charakteryzuje się poważnymi zaburzeniami neurologicznymi w pierwszym roku życia dziecka, w tym napadami padaczkowymi, zezem, hipertonicznością mięśni oraz opóźnieniami w rozwoju psychicznym i fizycznym. Często ta postać HD łączy się z wrodzoną rybią łuską. Choroba występuje u mniej niż 1 na 100 000 noworodków. Postępująca degeneracja psychomotoryczna kończy się śmiercią, zwykle związaną z niewydolnością oddechową.
− III typ (przewlekła neuropatia). Choroba typu 3, dawniej nazywana młodzieńczą chorobą Gauchera, charakteryzuje się powoli postępującym uszkodzeniem mózgu i poważnymi objawami w innych narządach. Ten typ choroby jest również bardzo rzadki. Oznaki i objawy choroby Gauchera typu 3 rozwijają się we wczesnym dzieciństwie i są podobne do objawów choroby typu 1, z wyjątkiem objawów zajęcia ośrodkowego układu nerwowego. Postawienie trafnego rozpoznania możliwe jest dopiero w przypadku progresji objawów neuropatii, potwierdzonej badaniami klinicznymi. Pacjenci z chorobą Gauchera typu 3, którzy osiągną wiek dorosły, mogą żyć dłużej niż 30 lat.
Diagnostyka (przychodnia)
DIAGNOSTYKA Ambulatoryjna
Kryteria diagnostyczne
Reklamacje i wywiad:
osłabienie, zwiększone zmęczenie;
· zwiększona podatność na infekcje (infekcje dróg oddechowych, bakteryjne);
· objawy zespołu krwotocznego (krwiaki podskórne, krwawienia z błon śluzowych) i/lub przedłużone krwawienie podczas drobnych zabiegów chirurgicznych;
· silny ból kości i stawów (charakter i lokalizacja bólu, historia złamań kości);
· opóźniony rozwój fizyczny i seksualny;
· objawy objawów neurologicznych (apraksja okoruchowa lub zez zbieżny, ataksja, utrata inteligencji, zaburzenia czucia itp.);
· wywiad rodzinny (obecność splenektomii lub wymienionych powyżej objawów u rodzeństwa, rodziców).
Zwiększona objętość brzucha
Badanie lekarskie:
· Generalna Inspekcja;
· Pomiar wzrostu, masy ciała, temperatury ciała;
· Ocena stanu układu kostno-stawowego;
· Identyfikacja objawów zespołu krwotocznego;
· Wykrywanie hepatosplenomegalii, limfadenopatii;
· Ocena skóry w okolicy stawów kolanowych i łokciowych (obecność/brak przebarwień).
Objawy kliniczne i przedmiotowe choroby Gauchera w zależności od wieku
| System | Objaw | Noworodki |
Dzieci do roku |
Dzieci | Nastolatki |
| OUN | Opóźnienie i regresja zdolności psychomotorycznych | - | +++ | ++ | ± |
| drgawki | - | +++ | ++ | ± | |
| Skóra | Skóra kolodionowa (obrzęk tylnej części stóp i dłoni) | +++ | - | - | - |
| Przewód pokarmowy | Hepatosplenomegalia | ++ | +++ | +++ | +++ |
| Marskość wątroby | - | - | - | - | |
| Okulistyczne | Nieprawidłowe ruchy oczu | - | +++ | ++ | ± |
| Hematologiczne | niedokrwistość | - | + | +++ | ++ |
| komórki piankowe | ++ | +++ | +++ | +++ | |
| pancytopenia | - | + | + | + | |
| małopłytkowość | - | + | +++ | +++ | |
| Szkieletowy | Ból kości | - | - | + | +++ |
| kifoza | - | - | ± | ++ | |
| osteoporoza | - | - | ± | ++ | |
| Złamania patologiczne | - | - | ± | + | |
| Oddechowy | Restrykcyjna choroba płuc, nadciśnienie płucne | - | ++ | ++ | + |
| Inny | Wczesna śmierć | +++ | +++ | ± | - |
| Specyficzne badania laboratoryjne | β-D-glukozydaza | ↓↓↓ | ↓↓ | ↓↓ | ↓↓ |
| Chitotriozydaza |
Badania laboratoryjne :
· Szczegółowe badanie krwi: małopłytkowość, leukopenia, niedokrwistość;
· BAK: podwyższony poziom enzymów we krwi – ALT, AST, badanie metabolizmu żelaza (żelazo w surowicy, TBSS, ferrytyna, transferyna) pomoże w diagnostyce różnicowej pomiędzy anemią przewlekłą a stanem niedoboru żelaza wymagającym standardowego leczenia;
· Oznaczanie aktywności enzymu glukocerebrozydazy i chitotriazydazy w zaschniętych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii – w celu potwierdzenia rozpoznania;
· Badania genetyki molekularnej w celu potwierdzenia diagnozy - identyfikacja genu glukocerebrozydazy zlokalizowanego na długim ramieniu chromosomu 1 (region 1q21q31);
· Badanie morfologiczne szpiku kostnego pozwala na identyfikację charakterystycznych elementów diagnostycznych – komórek Gauchera, a jednocześnie wyklucza rozpoznanie hemoblastozy lub choroby limfoproliferacyjnej jako przyczyny cytopenii i hepatosplenomegalii.
Studia instrumentalne
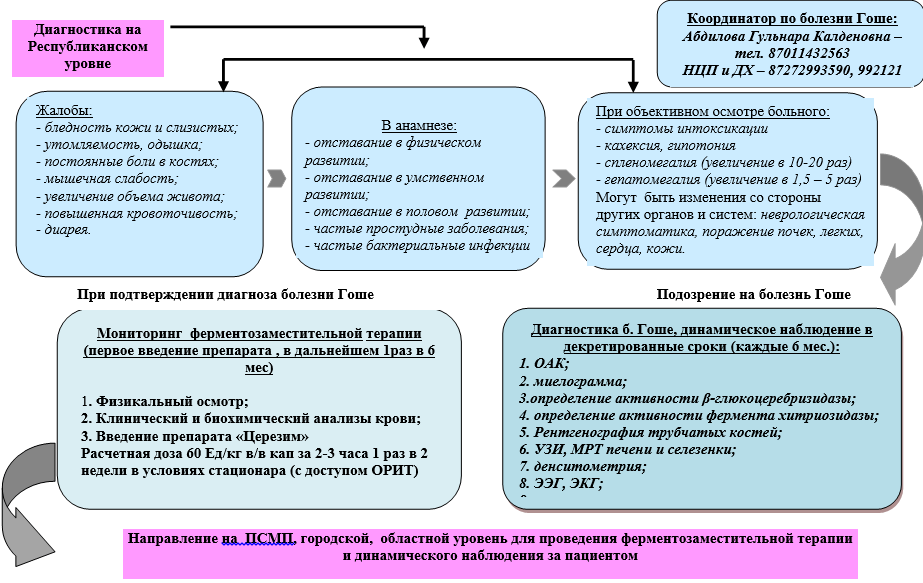
Algorytm diagnostyczny

Algorytm diagnostyki choroby Gauchera u dzieci na poziomie miasta i regionu

Algorytm diagnozowania choroby Gauchera u dzieci na poziomie republikańskim
Diagnostyka (szpital)
DIAGNOSTYKA NA POZIOMIE PACJENTA
Kryteria diagnostyczne:.
Algorytm diagnostyczny


Lista podstawowych środków diagnostycznych (UD - B)
szczegółowe badanie krwi
· chemia krwi
Oznaczanie aktywności enzymów glukocerebrozydazy i chitotriazydazy
· badania genetyki molekularnej
USG wątroby, śledziony
MRI kości udowej
· EKG
Rentgen kości szkieletowych
Lista dodatkowych środków diagnostycznych:
· Mielogram – badanie szpiku kostnego pozwala na wykrycie charakterystycznych elementów diagnostycznych – komórek Gauchera, a jednocześnie wyklucza rozpoznanie hemoblastozy lub choroby limfoproliferacyjnej jako przyczyny cytopenii i hepatosplenomegalii.
· Tomografia komputerowa płuc – w celu wykluczenia patologii układu oddechowego z przedłużającą się neutropenią.
· MRI mózgu – do diagnostyki różnicowej z chorobami onkologicznymi, z wyłączeniem uszkodzeń ośrodkowego układu nerwowego w długotrwałym zespole cytopenicznym (ryzyko udaru krwotocznego).
· MRI wątroby, śledziony – w przypadku hepatosplenomegalii istnieje duże ryzyko zawału wątroby i śledziony na skutek nacieku narządów i tkanek komórkami Gauchera.
· EchoCG – przy ciężkim tachykardii, na tle objawów niewydolności oddechowej z przedłużonym zespołem cytopenicznym, istnieje ryzyko powikłań ze strony układu sercowo-naczyniowego (wysiękowe zapalenie osierdzia, zapalenie mięśnia sercowego, dysfunkcja autonomiczna).
· Koagulogram – w przypadku stanu cytopenicznego, dodatku infekcji bakteryjnej lub wirusowej istnieje ryzyko wystąpienia stanu krwotocznego, stanu septycznego, zespołu rozsianego wykrzepiania wewnątrznaczyniowego.
· Dopplerografia naczyń układu wrotnego – w celu wykluczenia nadciśnienia wrotnego.
Powikłania infekcyjne na tle długotrwałego zespołu cytopenicznego towskazania do dodatkowych badań laboratoryjnych:
· badanie bakteriologiczne płynów biologicznych,
· badania serologiczne (wirusowe) w kierunku CMV, wirusowego zapalenia wątroby typu B, C, (D), HIV, EBV,
· oznaczenie białka C-reaktywnego (ilościowe),
· w przypadku wzrostu poziomu transaminaz: przeprowadzić badania serologiczne (wirusowe) w celu wykluczenia wirusowego zapalenia wątroby: CMV, A, B, C, EBV, jeśli wyniki PCR są pozytywne
· koagulogram – badanie hemostazy z ryzykiem powikłań septycznych, obfitego zespołu krwotocznego
· RTG kości szkieletowych – w celu identyfikacji i oceny stopnia zaawansowania uszkodzeń układu kostno-stawowego (rozlana osteoporoza, charakterystyczne kolbowate zniekształcenia dalszej kości udowej i bliższej kości piszczelowej (kolba Erlenmeyera), ogniska osteolizy, osteoskleroza i martwica kości, patologiczne złamania);
· Densytometria i rezonans magnetyczny (MRI) są metodami bardziej czułymi – pozwalają wykryć uszkodzenia kości (osteopenia, nacieki szpiku kostnego) we wczesnych stadiach, które nie są uwidocznione radiologicznie;
· USG i MRI wątroby i śledziony pozwalają na identyfikację ich zmian ogniskowych i określenie początkowej objętości narządów, co jest niezbędne do późniejszego monitorowania skuteczności enzymatycznej terapii zastępczej;
· Echokardiografia dopplerowska – u pacjentów po splenektomii;
esophagogastroduodenoskopia - w obecności odpowiednich dolegliwości lub objawów nadciśnienia wrotnego.
Diagnostyka różnicowa
Diagnostyka różnicowa
Chorobę Gauchera należy różnicować ze wszystkimi chorobami, którym towarzyszy powiększenie wątroby i śledziony, cytopenia, krwawienie i uszkodzenie kości.
| Diagnoza | Uzasadnienie diagnostyki różnicowej | Ankiety | Kryteria wykluczenia diagnozy |
| Hemoblastozy i chłoniaki | Krwotoczny sm, ból kości, hepatosplenomegalia, |
2.mielogram, |
|
| Nabyta niedokrwistość aplastyczna | Krwotoczny sm, (+/_) ból kości, pancytopenia |
1. Pełna morfologia krwi z liczbą płytek krwi, liczbą retikulocytów, 2.mielogram, 3.molekularne genetyczne badanie krwi |
1. Brak spadku aktywności enzymu glukocerebrozydazy i wzrost aktywności enzymu chitotriazydazy (w wysuszonych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii); 2. nie zidentyfikowano genu glukocerebrozydazy zlokalizowanego na długim ramieniu chromosomu 1 (region 1q21q31); 3. Podczas zliczania komórek na mielogramie nie wykryto komórek Gauchera |
| Przewlekłe cholestatyczne choroby wątroby, marskość wątroby w wyniku przewlekłego wirusowego i niewirusowego zapalenia wątroby | Hepatosplenomegalia, podwyższony poziom transaminaz, bilirubiny, sm cytopeniczny, sm krwotoczny, sm bolesny |
1. Pełna morfologia krwi z liczbą płytek krwi, liczbą retikulocytów, 2.mielogram, 3.molekularne genetyczne badanie krwi 4. oznaczanie aktywności enzymów glukocerebrozydazy i chytriozydazy 5.B/x badanie krwi 6. USG, CT, MRI narządów jamy brzusznej |
1. Brak spadku aktywności enzymu glukocerebrozydazy i wzrost aktywności enzymu chitotriazydazy (w wysuszonych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii); 2. nie zidentyfikowano genu glukocerebrozydazy zlokalizowanego na długim ramieniu chromosomu 1 (region 1q21q31); |
| Przewlekłe zapalenie kości i szpiku, gruźlica kości | Ossalgia, ograniczenie ruchomości kończyn |
2.mielogram, 3.molekularne genetyczne badanie krwi 4. oznaczanie aktywności enzymów glukocerebrozydazy i chytriozydazy 5.B/x badanie krwi |
1. Brak objawów cytopenii (spadek poziomu hemoglobiny, płytek krwi, leukopenia), 2. Brak spadku aktywności enzymu glukocerebrozydazy i wzrost aktywności enzymu chitotriazydazy (w wysuszonych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii); 3. nie zidentyfikowano genu glukocerebrozydazy zlokalizowanego na długim ramieniu chromosomu 1 (region 1q21q31); 4.brak choroby krwotocznej, 5. Charakterystyczne obrzęki kości piszczelowej w kształcie maczugi lub kolby („kolby Erlenmeyera”) nie są wykrywane na zdjęciu rentgenowskim. 5. Brak hepatosplenomegalii |
| Inne dziedziczne enzymopatie (choroba Niemanna-Picka |
Wczesny początek choroby (3-5 miesięcy), zwiększyć objętość brzucha, opóźniony rozwój psychomotoryczny, drgawki, inne objawy neurologiczne, bóle brzucha, krwawienia, niestabilność emocjonalna |
1.. Pełna morfologia krwi z liczbą płytek krwi, retikulocytów, 2.mielogram, 3. molekularne badania genetyczne krwi (oznaczenie mutacji w genach SMPD1, NPC1 i NPC2, genie glukocerebrozydazy, zlokalizowanych na długim ramieniu chromosomu 1 (region 1q21q31). 4. oznaczanie aktywności enzymów glukocerebrozydazy i chytriozydazy, sfingomielinazy 5.B/x badanie krwi 6. USG, CT, MRI narządów jamy brzusznej 7. Badanie rentgenowskie tkanki kostnej (R, MRI, CT) 8.Badanie przez neurologa |
1. Brak spadku aktywności enzymu glukocerebrozydazy i wzrost aktywności enzymu chitotriazydazy (w wysuszonych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii); |
| Histiocytoza | Ossalgia, ograniczona ruchomość kończyn, pancytopenia, sm krwotoczny, hepatosplenomegalia, zapalenie płuc, podatność na infekcje |
1. Pełna morfologia krwi z liczbą płytek krwi, liczbą retikulocytów, 2.mielogram, immunofenotyp szpiku kostnego 3.molekularne genetyczne badanie krwi 4. oznaczanie aktywności enzymów glukocerebrozydazy i chytriozydazy 5. Badanie krwi 6. USG, CT, MRI narządów jamy brzusznej 7. Badanie rentgenowskie tkanki kostnej (R, MRI, CT) |
1. Brak spadku aktywności enzymu glukocerebrozydazy i wzrost aktywności enzymu chitotriazydazy (w wysuszonych plamach krwi metodą tandemowej spektrometrii mas lub fluorymetrii); 2. nie wykryto genu glukocerebrozydazy zlokalizowanego na długim ramieniu chromosomu 1 (region 1q21q31); 3. Charakterystyczne obrzęki kości piszczelowej w kształcie maczugi lub kolby („kolby Erlenmeyera”) nie są wykrywane na zdjęciu rentgenowskim. |
Leczenie za granicą
Skorzystaj z leczenia w Korei, Izraelu, Niemczech i USA
Leczenie za granicą
Uzyskaj poradę dotyczącą turystyki medycznej
Leczenie
Leki (składniki aktywne) stosowane w leczeniu
| Azytromycyna |
| Alfakalcydol |
| Amfoterycyna B |
| Acyklowir |
| Wankomycyna |
| Worykonazol |
| Gentamycyna |
| Diklofenak |
| Ibuprofen |
| Imigluceraza |
| Immunoglobulina G |
| Jodiksanol |
| Kaspofungina |
| Klindamycyna |
| Kolekalcyferol |
| Laktuloza |
| Lornoksykam |
| Meropenem |
| Metronidazol |
| Mikafungina |
| Kompleks osseina-hydroksyapatyt |
| Paracetamol |
| Tramadol |
| Flukonazol |
| Cefotaksym |
| Ceftazydym |
| Ceftriakson |
Leczenie (przychodnia ambulatoryjna)
LEczenie ambulatoryjne
Taktyka leczenia
Pacjenci ze wszystkimi typami (I, II, III) choroby Gauchera leczeni są w trybie ambulatoryjnym.
Leczenie niefarmakologiczne:
· Schemat – leczniczo-ochronny w okresie zespołu cytopenicznego, krwotocznego, powikłań kostnych;
· Profilaktyka urazów, rehabilitacja przewlekłych ognisk infekcji;
· Korekta psychologiczna – psychoterapia, adaptacja psychologiczna.
Farmakoterapia
Nowoczesne leczenie GD polega na przepisywaniu dożywotniej enzymatycznej terapii zastępczej (ERT) z użyciem rekombinowanej glukocerebrozydazy, która łagodzi główne objawy kliniczne choroby, poprawiając jakość życia pacjentów z GD i nie powodując znaczących skutków ubocznych. . Każdemu pacjentowi z klinicznymi objawami GD (GD typu 1, GD typu 3) należy przepisać ERT. Dawkę leku należy dobierać indywidualnie, kierując się parametrami klinicznymi i laboratoryjnymi. W związku z rozwojem diagnostyki laboratoryjnej, badając rodzeństwo (brata i siostry probanda), można zidentyfikować dzieci z HD, które nie mają objawów klinicznych. Takich pacjentów należy monitorować, lecz leczenie należy rozpocząć dopiero w momencie pojawienia się objawów choroby.
ERT ma na celu dostarczenie wystarczającej ilości enzymu, która umożliwi rozkład złogów zbędnych substancji. Zatem enzymatyczna terapia zastępcza działa poprzez uzupełnienie lub zastąpienie brakującego lub wadliwego enzymu u pacjentów z chorobą Gauchera.

Lista niezbędnych leków
Imigluceraza
Leczenie patogenetyczne choroby Gauchera polega na stosowaniu przez całe życie enzymatycznej terapii zastępczej rekombinowaną glukocerebrozydazą. Początkowa dawka imiglucerazy na wstrzyknięcie dla GD typu I wynosi 30-40 jednostek/kg bez zmian kostnych i 60 jednostek/kg w przypadku obecności zmian kostnych. W przypadku typu III u dzieci dawka może dochodzić do 100-120 jednostek/kg .
Lek podaje się dożylnie w odstępach 1 raz na 2 tygodnie. (2 razy w miesiącu).
Możliwe jest stopniowe zmniejszanie dawki o 10-20 jednostek/kg z wyraźną dodatnią dynamiką po 1 roku leczenia GD typu 1 bez uszkodzeń kości i po 3-4 latach z początkowym uszkodzeniem szkieletu. Leczenie podtrzymujące: 15-60 jednostek/kg dożylnie, 3 godziny co 2 tygodnie, przez całe życie.
Protokół enzymatycznej terapii zastępczej imiglucerazą

Lista dodatkowych leków
· Paracetamol
Lornoksykam
Diklofenak
· Tramadol
Alfakalcydol
Flukanazol
Wapń Dz
Osteogenon
Acyklowir
Laktuloza
· Cefotaksym
· Ceftazydym
· Ceftriakson
Azytromycyna
· Gentamycyna
· Jodiksanol
Meropenem
Niesteroidowe leki przeciwzapalne:
· Paracetamol – tabletki 200 mg, 500 mg; świece. Dorośli: 500 mg 3-4 razy dziennie przez 3-7 dni. Dzieci w dawce 60 mg/kg/dzień w 3-4 dawkach, 3-7 dni;
· Ibuprofen tabletki 200 mg, 400 mg; Dzieci – ibuprofen 30-40 mg/kg/dzień,
· Lornaxicam – tabletki powlekane 4 mg, 8 mg. Dorośli: 8 mg 2 razy dziennie doustnie przez 2 tygodnie; liofilizat do sporządzania roztworu do podawania dożylnego i domięśniowego, 8 mg. Dorośli: 8 mg 2 razy dziennie, domięśniowo, 10 dni;
· Diklofenak – roztwór do wstrzykiwań 2,5% w ampułkach 3 ml, tabletki 0,05 g, tabletki opóźniające 0,025; 0,05 i 0,1 g; drażetki 0,025 g. Czopki doodbytnicze 0,05 i 0,1 g. Żel, krem, emulżel (1 g - 0,01 g ortofenu) w tubkach. Dzieci: 2-3 mg/kg/dzień, domięśniowo, przez 1-3-5 dni. Dorośli: 7 mg 2 razy dziennie domięśniowo przez 1-3-5 dni.
· Tramadol – roztwór do wstrzykiwań 50 mg/ml, czopki doodbytnicze 0,1 g, krople -2,5 mg/kroplę, kapsułki 50 mg. Doustnie, zwykle stosowana dawka początkowa dla dorosłych i dzieci powyżej 14 roku życia wynosi 50 mg (ponownie, jeśli nie ma efektu, po 30-60 minutach). Pozajelitowo (IV, IM, SC) - 50-100 mg, doodbytniczo - 100 mg (ponowne wprowadzenie czopków możliwe jest po 4-8 godzinach). Maksymalna dawka dobowa wynosi 400 mg (w wyjątkowych przypadkach można ją zwiększyć do 600 mg). Dzieci w wieku od 1 do 14 lat doustnie (krople) lub pozajelitowo – pojedyncza dawka 1-2 mg/kg, maksymalna dawka dobowa – 4-8 mg/kg.
Korektory metabolizmu tkanki kostnej i chrzęstnej:
· Alfakalcydol, kapsułki 0,5 µg. Dzienna dawka dla dorosłych waha się od 0,07 mcg do 20 mcg, dla dzieci 0,01-0,08 mcg/kg, dzienna dawka dla dzieci 0,01-0,08 mcg/kg.
· Calcium D3 - tabletki do żucia zawierające (składniki aktywne): węglan wapnia – 1250 mg (co odpowiada 500 mg wapnia pierwiastkowego); cholekalcyferol – 200 j.m. (jednostki międzynarodowe). Dorośli i dzieci powyżej 12. roku życia – 2 tabletki dziennie, najlepiej podczas posiłku.
· Osteogenon – tabletki kompleksu osseina-hydroksyapatyt – 830 mg; 2-4 tabletki x 2 razy dziennie.
Algorytm postępowania w sytuacjach awaryjnych

Interwencja chirurgiczna: NIE.
Inne rodzaje leczenia:
· resocjalizacja psychospołeczna: psychoterapia, adaptacja psychologiczna, terapia środowiskowa;
· adaptacja społeczna i poprawa jakości życia.
Wskazania do konsultacji ze specjalistami :
| Specjalista | Wskazanie |
| traumatolog - ortopeda |
Z wyłączeniem obecności patologii szkieletu u dziecka |
| Neuropatolog, psychoneurolog | ocena stanu neurologicznego, stanu neuropsychicznego, określenie rodzaju choroby |
| fizjoterapeuta |
określenie metod leczenia fizjoterapeutycznego |
| lekarz fizjoterapii | dobór indywidualnego programu fizjoterapeutycznego |
| genetyk | potwierdzenie diagnozy, genotypowanie |
| W razie potrzeby możliwa jest konsultacja z innymi specjalistami w zależności od przypadku klinicznego. | |
Działania zapobiegawcze:
· wczesna diagnostyka objawów klinicznych choroby Gauchera w celu zapobiegania powikłaniom;
· medyczne doradztwo genetyczne w celu wyjaśnienia ryzyka genetycznego.
· zapobieganie powikłaniom infekcyjnym na tle długotrwałego zespołu cytopenicznego, który w niektórych przypadkach jest główną przyczyną, a w niektórych przypadkach nawet śmiercią pacjenta.
· pielęgnacja jamy ustnej: 6-10 razy dziennie, płukanie jamy ustnej roztworami dezynfekcyjnymi przeznaczonymi do leczenia błony śluzowej jamy ustnej. Dokładna, ale delikatna pielęgnacja zębów i dziąseł; ograniczenie używania nawet miękkich szczoteczek do zębów; preferuj prysznic doustny; w przypadku trombocytopenii lub wrażliwych błon śluzowych należy wykluczyć stosowanie szczoteczek do zębów, zamiast tego konieczne jest dodatkowe leczenie jamy ustnej środkami ściągającymi.
Jeśli pojawią się objawy zapalenia jamy ustnej, do podstawowej terapii należy dodać:
· Flukonazol – szacunkowa dawka 4-5 mg/kg na dzień, kapsułki 50 mg, 100 mg, 150 mg, roztwory do infuzji 2 mg/ml, żel do leczenia jamy ustnej r.o.
· Acyklowir – dawka obliczona 250 mg/m2 2 x 3 razy dziennie, tabletki 200 mg, roztwór do wstrzykiwań 250 mg, maść do stosowania zewnętrznego.
· W przypadku pojawienia się uszkodzeń błony śluzowej jamy ustnej: unikać stosowania szczoteczek do zębów
2) wraz z rozwojem rozległego martwiczego zapalenia jamy ustnej wskazane jest ogólnoustrojowe leczenie przeciwgrzybicze i przeciwbakteryjne:
· Cefotaksym, butelka 1 g do przygotowania roztworu. Dorośli 1-2g 2-3 razy dziennie dożylnie, domięśniowo przez 7-10 dni. Dzieci 50-100 mg/kg masy ciała/dzień, 2-4 razy dziennie, domięśniowo, dożylnie, 7-10 dni;
· Ceftazydym, butelka 250 mg, 500 mg, 1 g, 2 g do przygotowania roztworu. Dorośli: 1-6 g/dzień w 2 lub 3 dawkach dożylnie lub domięśniowo. Dzieci powyżej 2 miesiąca życia: 30-100 mg/kg/dobę w 2-3 dawkach podzielonych, przy obniżonej odporności - do 150 mg/kg/dobę (maksymalnie 6 g/dobę) w 3 dawkach podzielonych. Noworodki i niemowlęta do 2 miesiąca życia: 25-60 mg/kg/dobę w 2 dawkach podzielonych.
· Ceftriakson, butelka 500 mg, 1 g do przygotowania roztworu. Dzieci 50-80 mg/kg/dzień krople dożylne 1 godzina przez 7-10 dni;
· Jodiksanol, roztwór do wstrzykiwań, 100 mg/2 ml i 500 mg/2 ml. Dorosłym i dzieciom powyżej 12. roku życia przepisuje się domięśniowo, dożylnie (strumień, przez 2 minuty lub kroplówkę) w dawce 5 mg/kg co 8 godzin lub 7,5 mg/kg co 12 godzin przez 7-10 dni.
· Gentamycyna, roztwór do wstrzykiwań, ampułki 40 mg/ml. dorośli 3-5 mg/kg (maksymalna dawka dobowa) w 3-4 dawkach przez 7-10 dni. Jest przepisywany małym dzieciom wyłącznie ze względów zdrowotnych w przypadku ciężkich infekcji. Maksymalna dawka dobowa dla dzieci w każdym wieku wynosi 5 mg/kg.
· Azytromycyna, kapsułki 250, 500 mg. Dla dzieci o masie ciała powyżej 10 kg w dawce: w 1. dniu – 10 mg/kg masy ciała; w ciągu kolejnych 4 dni – 5 mg/kg. Możliwy jest 3-dniowy cykl leczenia; w tym przypadku pojedyncza dawka wynosi 10 mg/kg. (Dawka kursu 30 mg/kg masy ciała). Dorośli z infekcjami górnych i dolnych dróg oddechowych, infekcjami skóry i tkanek miękkich przepisuje się 0,5 g pierwszego dnia, następnie 0,25 g od 2 do 5 dnia lub 0,5 g dziennie przez 3 dni (dawka kursowa 1,5 g ).
· Meropenem, proszek do sporządzania roztworu do podawania dożylnego, 0,5 i 1,0 g. Dla dzieci w wieku od 3 miesięcy do 12 lat zalecana dawka wynosi 10-20 mg/kg co 8 godzin, w zależności od rodzaju i ciężkości zakażenia, wrażliwości patogenu i stanu pacjenta. U dzieci o masie ciała powyżej 50 kg należy stosować dawkę dla dorosłych.
3) Dekontaminację jelit przeprowadza się według uznania szpitala, można odmówić wykonania dekontaminacji. W przypadku początkowych zmian jelitowych zaleca się dekontaminację (leczenie zapobiegawcze). Do selektywnej dekontaminacji jelit:
Cyprofloksacyna w dawce 20 mg/kg dziennie, 100 mg w butelce, 250 mg, 500 mg w tabletkach, krople do oczu, krople do uszu;
4) Wszystkie osoby opiekujące się pacjentem – rodzice i osoby odwiedzające – mają obowiązek utrzymywać higienę osobistą i stale myć ręce.
Taktyka terapii substytucyjnej oraz zgodnie z Zarządzeniem nr 666 „Po zatwierdzeniu Nomenklatury, Regulaminu pobierania, przetwarzania, przechowywania, sprzedaży krwi oraz Regulaminu przechowywania, przetaczania krwi, jej składników i produktów krwiopochodnych z dnia 6 marca 2011 roku, Załącznik do zarządzenia nr 417 z dnia 29 maja 2015 r.
Monitorowanie pacjenta:
· ERT przez całe życie;
kontrola dynamiczna: 1 rok – raz na 3 miesiące, następnie raz na 6 miesięcy:
· adaptacja społeczna;
· obserwacja genetyka rodziny pacjenta HD.
Wskaźniki skuteczności leczenia:
· poprawa/stabilizacja parametrów hematologicznych (ustąpienie zespołu cytopenicznego, brak uzależnienia od transfuzji krwi);
· przywrócenie poziomu glukocerebrozydazy, obniżenie poziomu chitotriozydazy;
· eliminacja bólu;
· odbudowa tkanki kostnej;
· poprawa/stabilizacja pracy narządów pozabrzusznych (serce, płuca, oczy);
· zmniejszenie częstości infekcji dróg oddechowych;
· zmniejszenie tempa postępu choroby;
poprawa jakości życia pacjenta (przywrócenie rozwoju psychicznego, duchowego, fizycznego).
Leczenie (pogotowie)
DZIAŁANIA DIAGNOSTYCZNE PROWADZONE NA ETAPIE OPIEKI NAGŁYCH
Środki diagnostyczne:
· wykonanie wywiadu;
· badanie lekarskie;
· określenie patologii serca (pulsoksymetria, ciśnienie krwi, częstość akcji serca, EKG).
Farmakoterapia
Resuscytacja krążeniowo-oddechowa zgodnie ze wskazaniami;
· terapia syndromowo-objawowa według wskazań;
· Terapia tlenowa;
· zapobieganie aspiracji;
· przeciwbólowa terapia przeciwzapalna
Leczenie (szpitalne)
LECZENIE SZACUNKOWE
Taktyka leczenia:patrz poziom ambulatoryjny.
Farmakoterapia: zobacz poziom ambulatoryjny.
W przypadku poważnych powikłań leczenie farmakologiczne prowadzi się zgodnie z Protokołami klinicznymi.
Farmakoterapię intensyfikuje się w przypadku wystąpienia powikłań na tle długotrwałego zespołu cytopenicznego, nawarstwiania się infekcji wirusowo-bakteryjnej lub progresji choroby podstawowej. Najpoważniejszymi powikłaniami zagrażającymi życiu są powikłania infekcyjne. Obecność gorączki u pacjenta z neutropenią (neutrofile< 500/мкл) считается однократное повышение температуры тела >37,9 0 Trwający dłużej niż godzinę lub kilka wzrostów (3 - 4 razy dziennie) do 38 0 C. Biorąc pod uwagę duże ryzyko śmiertelnej infekcji, gorączkę u pacjenta z neutropenią uważa się za obecność infekcji , co nakazuje natychmiastowe rozpoczęcie empirycznej terapii przeciwbakteryjnej i przeprowadzenie badania w celu wyjaśnienia charakteru zakażenia. Zaproponowano wiele początkowych schematów leczenia przeciwbakteryjnego, których skuteczność jest zasadniczo identyczna.
Postanowienia ogólne:
· przy wyborze wyjściowej kombinacji antybiotyków należy wziąć pod uwagę: wyniki powtarzanych w tej klinice badań bakteriologicznych u innych pacjentów; czas trwania aktualnej neutropenii, historia infekcji pacjenta, poprzednie kursy antybiotyków i ich skuteczność
· wraz z pojawieniem się gorączki wszystkie inne dane kliniczne: podciśnienie tętnicze, niestabilna hemodynamika są wskazaniem do natychmiastowego przepisania kombinacji antybiotyków: karbopenemy (meropenem (lub imipenem/cylastatyna)) + aminoglikozyd (amikacyna) + wankomycyna.
· długo utrzymujące się CVC i gorączka po płukaniu i/lub nie tylko gorączka, ale niesamowite dreszcze ®Wankomycyna jest już w kombinacji wyjściowej;
· klinika zapalenia jelit z biegunką: do połączenia początkowego – wankomycyna per os 20 mg/kg dziennie. Można przepisać Metronidazol (doustnie i/lub dożylnie)
· ciężkie zapalenie jamy ustnej ze zmianami zapalnymi dziąseł ® penicylina, klindamycyna w połączeniu z beta-laktamem lub meropenemem/
charakterystyczna wysypka i/lub obecność druz grzybiczych w moczu i/lub charakterystyczne zmiany w wątrobie i śledzionie podczas USG®
· Amfoterycyna B – liofilizat do sporządzania roztworu. Dawka początkowa wynosi 1. dnia 0,5 mg/kg, następnego dnia pełna dawka terapeutyczna wynosi 1 mg/kg dziennie jednorazowo. Podczas stosowania Amfoterycyny B należy monitorować czynność nerek oraz wykonać biochemiczne badanie krwi (elektrolity, kreatynina). Konieczna jest ciągła korekta potasu do wartości prawidłowych. Podczas infuzji amfoterycyny B, a także przez około 3-4 godziny po infuzji, można zaobserwować reakcje na podanie leku w postaci gorączki, dreszczy i tachykardii, które można złagodzić za pomocą leków przeciwbólowych. W przypadku zaburzeń czynności nerek konieczne jest stosowanie worykonazolu, Cancidas i lipidowych postaci amforecyny B.
· Worykonazol - tabletka 50 mg, liofilizat do sporządzania roztworu 200 mg/butelka SD 4-6 mg/kg.
Kaspofungina - liofilizat do sporządzania roztworu do infuzji 50 mg
Micofungin - liofilizat do sporządzania roztworu do infuzji 50 mg
Zmiana antybiotyków z uwzględnieniem wrażliwości izolowanej flory. Skuteczność wstępnej antybiotykoterapii należy ocenić po 72 godzinach, jednak zawsze konieczne jest szczegółowe badanie takiego pacjenta w odstępach 8-12 godzin, oceniające stabilność hemodynamiczną i stopień zatrucia oraz pojawienie się nowych ognisk infekcyjnych. Terapię antybakteryjną kontynuuje się do ustąpienia neutropenii i całkowitego ustąpienia wszystkich ognisk infekcyjnych.
W przypadku głębokiej aplazji ryzyko powikłań septycznych zmniejsza się po zastosowaniu biernej immunizacji immunoglobulinami G w dawce 0,1-0,2 g/kg/dzień dożylnie.
Lista niezbędnych leków:
Imigluceraza 30-60 jednostek/kg kropla dożylna przez 3 godziny
Lista dodatkowych leków:
· Paracetamol
Lornoksykam
Diklofenak
· Tramadol
Alfakalcydol
Flukanazol
Wapń Dz
Osteogenon
Acyklowir
Laktuloza
· Cefotaksym
· Ceftazydym
· Ceftriakson
Azytromycyna
· Gentamycyna
· Jodiksanol
Meropenem
Immunoglobulina G
Amfoterycyna B
Worykonazol
Kaspofungina
Mikofungina
Wankomycyna
Metronidazol
· Klindamycyna
Interwencja chirurgiczna:
· korekcja złamań patologicznych tkanki kostnej, przykurczów w stawie.
Inne rodzaje leczenia:
· rehabilitacja ruchowa: fizjoterapia, ćwiczenia lecznicze, masaże;
· resocjalizacja psychospołeczna: psychoterapia, adaptacja psychologiczna, terapia środowiskowa.
Wskazania do konsultacji ze specjalistami: zobacz poziom ambulatoryjny.
Wskazania do przeniesienia na oddział intensywnej terapii:
· niewyrównany stan pacjenta;
· uogólnienie procesu wraz z rozwojem powikłań wymagających intensywnego monitorowania i terapii;
· okres pooperacyjny;
· rozwój powikłań w trakcie intensywnej chemioterapii, wymagających intensywnego leczenia i obserwacji.
Wskaźniki skuteczności leczenia:
· przywrócenie rozwoju psychicznego, duchowego i fizycznego;
· przywrócenie mobilności i wydajności;
· eliminacja bólu w ciągu pierwszych 2 lat terapii;
· zapobieganie kryzysom kostnym;
· zapobieganie rozwojowi martwicy kości i zapadnięciu się tkanki podchrzęstnej;
· poprawa gęstości mineralnej kości;
· wzrost gęstości mineralnej kości w ciągu 3 lat terapii;
· osiągnięcie normalnego tempa wzrostu, zgodnie ze standardami populacji, w ciągu 3 lat terapii;
· osiągnięcie normalnego wieku dojrzewania.
· normalizacja morfologii krwi w ciągu pierwszych 3 lat terapii;
· zmniejszenie hepatosplenomegalii;
· poprawa stanu narządów pozabrzusznych (serce, płuca, oczy).
Dalsze zarządzanie:
Po ustabilizowaniu się stanu, przywróceniu parametrów hematologicznych, ustąpieniu bólu, zatrucia i objawów krwotocznych dziecko zostaje wypisane do leczenia ambulatoryjnego pod kontrolą pediatry lub miejscowego hematologa w celu kontynuacji enzymatycznej terapii zastępczej pod nadzorem badań. Dalsze monitorowanie stanu pacjenta odbywa się na poziomie ambulatoryjnym.
Hospitalizacja
Wskazania do planowej hospitalizacji
Wskazana jest planowa hospitalizacja w szpitalu w celu weryfikacji rozpoznania i dostosowania dawki enzymatycznej terapii zastępczej.
Wskazania do pilnej hospitalizacji
· Zespół cytopeniczny;
· Zespół silnego bólu („kryzys kostny”);
· Złamanie patologiczne kości szkieletowych;
· Niewydolność oddechowa.
Informacja
Źródła i literatura
- Protokoły z posiedzeń Komisji Wspólnej ds. Jakości Usług Medycznych Ministerstwa Zdrowia Republiki Kazachstanu, 2016
- 1) Zub N.V. „Choroba Gauchera: częstość występowania, semiotyka, jakość życia oraz kliniczne i ekonomiczne uzasadnienie enzymatycznej terapii zastępczej” streszczenie autorstwa dr hab. Moskwa 2010 2) Lukina E.A. „Choroba Gauchera: aktualny stan zagadnienia” Russian Medical News 2008, tom XIII, nr 2, s. 10-12. 51-56. 3) Belogurova M.B. „Patogeneza, obraz kliniczny, diagnostyka i leczenie choroby Gauchera”. Pediatria i chirurgia dziecięca. Nr 3 2010, s. 43-48. 4) Aerts J.M., van Weely S., Broot R. i in. Patogeneza zaburzeń spichrzania lizosomalnego na przykładzie choroby Gauchera // J. Inher. Metab. Dis. – 1993. – Cz. 16. nr 2 – s. 288-291. 5) Beutler E., Grabowski G.A., Scriver C.R. i in. Metaboliczne i molekularne podstawy chorób dziedzicznych //McGraw-Hill, Nowy Jork, 2001. – P.3635-3668. 6) de Frost M., vom Dahl S., Weverling G.J. i in. Zwiększona zapadalność na nowotwory u dorosłych Choroba Gauchera w Europie Zachodniej // Komórki krwi Mol. Dis. – 2006. – Cz. 36. – s. 53-58. 7) Taddei T.H., Kacena K.A., Yang M. i in. Niedoceniany postępujący charakter choroby Gauchera N370S i ocena ryzyka raka u 403 pacjentów // Am. J. Hematol. – 2009. – Cz. 84. nr 4. – s. 208-214. 8) Choroba Niederau C. Gauchera. Brema:UNI-MED; 2006. 84 s. 9) Zimran A., Kay A., Beutler E. i in. Choroba Gauchera: cechy kliniczne, laboratoryjne, radiologiczne i genetyczne 53 pacjentów. Medycyna 1992; 71: 337–53. 10) Weinreb N. J. Choroba Gauchera typu I u pacjentów w podeszłym wieku. Gauchera Clina. Persp. 1999; 7 ust. 2: 1–8. 11) Vorobyov A.I. (red.) Racjonalna farmakoterapia chorób układu krwionośnego. M.: Littera, 2009, 563–6. 12) AV Davydova „Lizosomalne choroby spichrzeniowe: choroba Gauchera” Siberian Medical Journal, 2009, nr 5. Str. 9-14. 13) Mikosch P., Reed M., Baker R. i in. Zmiany metabolizmu kości u siedmiu pacjentów z chorobą Gauchera leczonych kolejno imiglucerazą i miglustatem // Calcif. Tkanina Int. – 2008. – Cz. 83, nr 1. – s. 43-54. 14) vom Dahl S., Poll L., Di Rocco M. i in. Oparte na dowodach zalecenia dotyczące monitorowania chorób kości i odpowiedzi na enzymatyczną terapię zastępczą u pacjentów z chorobą Gauchera // Current Med. Badania i opinia. – 2006. – Cz. 22. nr 6. – s. 1045-1064. 15) Wenstrup R.J., Roca-Espiau M., Weinreb N.J. i in. Szkieletowe aspekty choroby Gauchera: recenzja // Br. J. Radiol. – 2002. – Cz. 75. – s. 2-12. 16) Cox TM, Schofield JP. Choroba Gauchera: cechy kliniczne i historia naturalna. Hematologia kliniczna Bailliere’a. 1997;10(4): 657-689. 17) Choroba Grabowskiego G. Gauchera: Enzymologia, genetyka i leczenie. W: Harris H., Hirshchorn K., wyd. Postępy w genetyce człowieka. Nowy Jork, Nowy Jork: Plenum Press; 1993;21:377-441. 18) Vorobyov A.I. (red.) Racjonalna farmakoterapia chorób układu krwionośnego. M.: Littera, 2009, 563–6. 19) Panel Oceny Technologii NIH ds. choroby Gauchera Choroba Gauchera: aktualne problemy w diagnostyce i leczeniu JAMA. 1996;275:548-553. Panel Oceny Technologii NIH ds. choroby Gauchera Choroba Gauchera: aktualne problemy w diagnostyce i leczeniu JAMA. 1996;275:548-553. 20) Grabowski G. A. Fenotyp, diagnostyka i leczenie choroby Gaushera // Lancet.-2008.- Vol. 372.Nr 9645.-R. 1263-1271. 21) Abdilova G.K., Boranbaeva R.Z., Omarova K.O. i in. Zalecenia metodyczne „Nowoczesna diagnostyka i leczenie choroby Gauchera u dzieci w Kazachstanie”, Almaty 2015 s. 26-27. 22). Przewodnik dla lekarzy dotyczący diagnostyki, leczenia i monitorowania dziedzicznych chorób metabolicznych, wyd. N.Blau, M.Duran, K.M. Gibson, C. Dionisi-Vici. 2014) 23) „Federalne wytyczne kliniczne dotyczące zapewnienia opieki medycznej dzieciom z chorobą Gauchera” Moskwa, 2015
Informacja
SKRÓTY UŻYTE W PROTOKOLE:
ALT – aminotransferaza alaninowa
AST – aotokolaminotransferaza asparaginowa
GD – choroba Gauchera
MRI – rezonans magnetyczny
CBC – pełna morfologia krwi
OAM - ogólna analiza moczu
USG – badanie USG
ERT – enzymatyczna terapia zastępcza
EKG - elektrokardiogram
EchoCG - echokardiografia
LSD – lizosomalne choroby spichrzeniowe
OUN – centralny układ nerwowy
DNA - kwas dezoksyrybonukleinowy
HS - zespół krwotoczny
ESR – szybkość sedymentacji erytrocytów
Tomografia komputerowa CT
LISTA TWÓRCÓW PROTOKOŁU:
1) Boranbaeva Riza Zulkarnaevna – doktor nauk medycznych, dyrektor Republikańskiego Przedsiębiorstwa Państwowego „Centrum Naukowe Pediatrii i Chirurgii Dziecięcej”.
2) Gulnara Kaldenovna Abdilova – kandydat nauk medycznych, zastępca dyrektora Republikańskiego Przedsiębiorstwa Państwowego „Centrum Naukowe Pediatrii i Chirurgii Dziecięcej” w dziedzinie pediatrii.
3) Omarova Kulyan Omarovna – doktor nauk medycznych, profesor, główny badacz Republikańskiego Przedsiębiorstwa Państwowego „Centrum Naukowe Pediatrii i Chirurgii Dziecięcej”.
4) Manzhuova Lyazat Nurbapaevna - kandydat nauk medycznych, kierownik Katedry Onkohematologii dla Starszych Dzieci Republikańskiego Przedsiębiorstwa Państwowego „Centrum Naukowe Pediatrii i Chirurgii Dziecięcej”.
5) Elmira Maratovna Satbaeva – Kandydat nauk medycznych, RSE w PME „Kazachski Narodowy Uniwersytet Medyczny im. S.D. Asfendiyarowa”, kierownik Katedry Farmakologii.
OGŁOSZENIE O BRAK KONFLIKTÓW INTERESÓW: brakuje.
RECENZENCI:
1. Kurmanbekova Saule Kaspakovna – Profesor Katedry Staży i Rezydencji w Pediatrii nr 2 Kazachskiego Narodowego Uniwersytetu Medycznego. SD Afendiyarova.
WSKAZANIE WARUNKÓW REWIZJI PROTOKOŁU: rewizja protokołu 3 lata po jego wejściu w życie i/lub gdy staną się dostępne nowe metody diagnostyki/leczenia o wyższym poziomie dowodów.
Załączone pliki
Uwaga!
- Samoleczenie może spowodować nieodwracalne szkody dla zdrowia.
- Informacje zamieszczone w serwisie MedElement nie mogą i nie powinny zastępować bezpośredniej konsultacji z lekarzem. Jeśli masz jakiekolwiek niepokojące Cię choroby lub objawy, skontaktuj się z placówką medyczną.
- Wybór leków i ich dawkowanie należy omówić ze specjalistą. Tylko lekarz może przepisać odpowiedni lek i jego dawkowanie, biorąc pod uwagę chorobę i stan organizmu pacjenta.
- Strona internetowa MedElement ma wyłącznie charakter informacyjny i źródłowy. Informacje zamieszczone na tej stronie nie powinny być wykorzystywane do bezprawnej zmiany zaleceń lekarskich.
- Redaktorzy MedElement nie ponoszą odpowiedzialności za jakiekolwiek obrażenia ciała lub szkody majątkowe powstałe w wyniku korzystania z tej witryny.
Jest to rzadka choroba genetyczna, której skuteczność leczenia z reguły zależy od terminowej diagnozy i odpowiedniego leczenia.
Choroba Gauchera jest chorobą dziedziczną o podłożu genetycznym, zaliczaną do chorób spichrzeniowych. Podstawą choroby jest niedobór aktywności enzymu glukocerebroidazy.
W organizmie zdrowego człowieka enzym ten umożliwia przetwarzanie odpadów przemiany komórkowej, jednak przy jego niedoborze w komórkach narządów wewnętrznych gromadzi się glukocerebrozyd, organiczna substancja tłuszczowa. Proces ten po raz pierwszy opisał francuski lekarz Philippe Gaucher w 1882 roku, od którego wzięła się nazwa choroby.
Z reguły choroba Gauchera atakuje najpierw wątrobę i śledzionę, ale komórki akumulacyjne mogą pojawiać się także w innych narządach - w mózgu i szpiku kostnym, nerkach i płucach.
Przyczyny choroby Gauchera.
Istnieją różne informacje na temat tej konkretnej choroby, z reguły badacze twierdzą, że choroba ta występuje raz na kilkadziesiąt tysięcy przypadków. W Federacji Rosyjskiej choroba Gauchera znajduje się na liście chorób sierocych (rzadkich).
Choroba Gauchera typu 1 występuje częściej w grupie etnicznej Żydów aszkenazyjskich, może jednak pojawić się u osób innych grup etnicznych.
Przyczyną choroby jest proces mutacji genu glukocerebrozydu (w organizmie człowieka występują dwa geny). Kiedy jeden gen jest zdrowy, a drugi dotknięty, osoba staje się nosicielem choroby Gauchera.
Prawdopodobieństwo urodzenia się osoby z chorobą Gauchera, którego rodzice są klinicznie zdrowi, jest możliwe, jeśli zarówno matka, jak i ojciec są nosicielami uszkodzonego genu. Trudność polega na tym, że nosiciel genu nie doświadcza objawów choroby, czyli nie myśli o konieczności wykonania badań genetycznych.
Objawy i oznaki choroby Gauchera.
Objawy i przebieg choroby różnią się w zależności od rodzaju:
Najczęstsza jest choroba pierwszego typu: choroba może pojawić się w każdym wieku, czasami ma przebieg bezobjawowy i nie wpływa na układ nerwowy
Najrzadsze są typy 2 i 3 choroby: początkowe objawy występują w dzieciństwie, choroba wpływa na układ nerwowy i postępuje z czasem
Początek choroby objawia się bólem brzucha, osłabieniem i ogólnym dyskomfortem. W wyniku tego, że śledziona i wątroba jako pierwsze są dotknięte akumulacją komórek Gauchera, następuje wzrost ich wielkości, co przy braku skutecznego leczenia może wywołać dysfunkcję wątroby i pęknięcie śledziony.
Często obserwuje się patologię kości (zwykle u dzieci), a mianowicie kości szkieletowe są słabe i słabo się rozwijają, w wyniku czego prawdopodobne jest opóźnienie wzrostu.
Diagnostyka choroby Gauchera.
Mutację tę można wykryć za pomocą testu DNA na wczesnym etapie ciąży. U dorosłych i dzieci wykrycie choroby wymaga wykonania badania szpiku kostnego lub badania enzymów krwi.
Leczenie choroby Gauchera.
Leczenie tej choroby odbywa się w oparciu o enzymatyczną terapię zastępczą, która polega na systematycznym dożylnym podawaniu specjalnych leków, co pomaga wyeliminować objawy choroby Gauchera typu 1. Leczenie choroby Gauchera typu 2 i 3 jest trudniejsze i wymaga kompleksowej terapii.
Rokowanie w chorobie Gauchera.
Prognozę stanu zdrowia i przewidywanej długości życia osoby chorej na chorobę Gauchera może określić wyłącznie specjalista na podstawie kompleksowego badania.
Catad_tema Mukowiscydoza i inne enzymopatie - artykuły
ICD 10: E75.2
Rok zatwierdzenia (częstotliwość aktualizacji): 2016 (przegląd co 2 lata)
ID: KR124
Stowarzyszenia zawodowe:
- Krajowe Towarzystwo Hematologiczne
Zatwierdzony
Rosyjskie Stowarzyszenie Hematologów
Zgoda
Rada Naukowa Ministerstwa Zdrowia Federacji Rosyjskiej__ __________201_
Enzymatyczna terapia zastępcza
Glukocerebrozydaza
Enzymodiagnostyka
Hepatomegalia
Splenomegalia
Cytopenia
Martwica aseptyczna
Uszkodzenie kości
Lista skrótów
ERT – enzymatyczna terapia zastępcza
APTT – czas częściowej tromboplastyny po aktywacji
USG – badanie USG
MRI – rezonans magnetyczny
CT – tomografia komputerowa
Warunki i definicje
β-glukocerebrozydaza (β-glukozydaza)- enzym lizosomalny biorący udział w degradacji komórkowych produktów przemiany materii
Komórki Gauchera– makrofagi przeciążone lipidami, średnica około 70-80 µm, kształt owalny lub wielokątny z bladą, piankową cytoplazmą.
Kolby Erlenmeyera- deformacja dystalnych kości udowych w kształcie kolby, ujawniona w badaniu radiologicznym
Enzymodiagnostyka- metody diagnostyki chorób, stanów i procesów patologicznych oparte na oznaczaniu aktywności enzymów w płynach biologicznych.
Enzymatyczna terapia zastępcza(enzymatyczna terapia zastępcza) to metoda leczenia chorób genetycznych, które są wynikiem dysfunkcji biochemicznej spowodowanej zmniejszoną aktywnością enzymów.
1. Krótka informacja
1 . 1 Definicja
choroba Gauchera – najczęstsza postać rzadkich dziedzicznych enzymopatii, zrzeszona w grupie lizosomalnych chorób spichrzeniowych.
1.2 Etiologia i patogeneza
Choroba opiera się na dziedzicznym niedoborze aktywności β-glukocerebrozydazy (β-glukozydazy), enzymu lizosomalnego biorącego udział w degradacji komórkowych produktów metabolizmu.
Choroba Gauchera dziedziczona jest w sposób autosomalny recesywny. Choroba opiera się na mutacjach w genie glukocerebrozydazy, zlokalizowanym w regionie q21 na chromosomie 1. Obecności dwóch zmutowanych alleli genu towarzyszy spadek (<30%) каталитической активности глюкоцереброзидазы, что приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше) – перегруженных липидами макрофагов. Следствием данного метаболического дефекта являются:
Przewlekła aktywacja układu makrofagów;
Autokrynna stymulacja monocytopoezy i wzrost bezwzględnej liczby makrofagów w miejscach „fizjologicznego domu”: śledzionie, wątrobie, szpiku kostnym, co powoduje powiększenie śledziony, powiększenie wątroby, naciek szpiku kostnego;
Upośledzenie funkcji regulacyjnych makrofagów, co prawdopodobnie leży u podstaw zespołu cytopenicznego i uszkodzeń układu kostno-stawowego.
1.3 Epidemiologia
Choroba Gauchera występuje z częstością od 1:40 000 do 1:60 000 we wszystkich grupach etnicznych; w populacji Żydów aszkenazyjskich zapadalność na tę chorobę sięga 1:450.
1.4 Kodowanie według ICD 10
E75.2 – Inne sfingolipidozy
1.5 Klasyfikacja
W zależności od obecności lub braku uszkodzenia ośrodkowego układu nerwowego i jego cech charakterystycznych wyróżnia się trzy typy choroby Gauchera:
typ I- bez objawów neurologicznych, najczęstszy wariant choroby, obserwowany u 94% pacjentów z chorobą Gauchera;
typ II (ostra neuropatia)- występuje u małych dzieci, charakteryzuje się postępującym przebiegiem i poważnym uszkodzeniem ośrodkowego układu nerwowego, prowadzącym do śmierci (pacjenci rzadko przeżywają powyżej 2. roku życia);
typ III (przewlekła neuropatia)- skupia bardziej niejednorodną grupę pacjentów, u których powikłania neurologiczne mogą objawiać się zarówno we wczesnym, jak i młodzieńczym wieku.
Typ I jest najczęstszym wariantem klinicznym choroby Gauchera i występuje zarówno u dzieci, jak i dorosłych. Średni wiek pacjentów w momencie pojawienia się choroby wynosi od 30 do 40 lat. Spektrum objawów klinicznych jest bardzo szerokie: na jednym końcu znajdują się pacjenci „bezobjawowi” (10–25%), na drugim pacjenci o ciężkim przebiegu: masywne powiększenie wątroby i śledziony, głęboka niedokrwistość i trombocytopenia, silne wyczerpanie i ciężkie powikłania zagrażające życiu (krwotoki, zawały śledziony, zniszczenie kości). Pomiędzy tymi polarnymi grupami klinicznymi znajdują się pacjenci z umiarkowaną hepatosplenomegalią i prawie prawidłowym składem krwi, z uszkodzeniem kości lub bez. Dzieci doświadczają opóźnień w rozwoju fizycznym i seksualnym; Charakteryzuje się specyficznym przebarwieniem skóry w okolicy stawów kolanowych i łokciowych.
W przypadku choroby Gauchera typu II główne objawy pojawiają się w ciągu pierwszych 6 miesięcy życia. We wczesnych stadiach choroby obserwuje się hipotonię mięśni, opóźnienie i regresję rozwoju psychomotorycznego. W miarę postępu choroby pojawia się spastyczność z charakterystycznym cofnięciem szyi i zgięciem kończyn, zaburzenia okoruchowe z rozwojem zeza zbieżnego, skurczem krtani i dysfagią. Charakterystyczne są zaburzenia opuszkowe z częstymi aspiracjami, prowadzące do śmierci pacjenta z powodu bezdechu, zachłystowego zapalenia płuc lub dysfunkcji ośrodka oddechowego mózgu. W późniejszych stadiach rozwijają się napady toniczno-kloniczne, które są zwykle oporne na leczenie przeciwdrgawkowe. Choroba jest śmiertelna w pierwszym lub drugim roku życia dziecka.
W przypadku choroby Gauchera typu III Objawy neurologiczne pojawiają się później, zwykle w wieku od 6 do 15 lat. Charakterystycznym objawem jest niedowład mięśni unerwionych przez nerw okoruchowy. Można zaobserwować drgawki miokloniczne i uogólnione toniczno-kloniczne, pojawia się i postępuje sztywność pozapiramidowa, obniżona inteligencja, szczękościsk, grymasy twarzy, dysfagia i skurcz krtani. Stopień upośledzenia umysłowego jest różny, od drobnych zmian osobowości po ciężką demencję. Można zaobserwować zaburzenia móżdżku, zaburzenia mowy i pisania, zmiany w zachowaniu i epizody psychozy. W większości przypadków przebieg choroby jest powoli postępujący. Śmierć następuje z powodu ciężkiego uszkodzenia płuc i wątroby. Oczekiwana długość życia pacjentów z chorobą Gauchera typu III może sięgać 12-17 lat, w pojedynczych przypadkach - 30-40 lat.
1.6 Objawy kliniczne
Główne objawy kliniczne choroby Gauchera obejmują powiększenie śledziony, powiększenie wątroby, cytopenię i zmiany kostne.
Splenomegalia- śledziona może być powiększona 5-80 razy w porównaniu do normalnej. W miarę postępu splenomegalii w śledzionie mogą rozwinąć się zawały, które z reguły nie mają objawów klinicznych.
Hepatomegalia- wielkość wątroby zwykle zwiększa się 2-4 razy. W badaniu USG można wykryć ogniskowe zmiany w wątrobie, które najprawdopodobniej wynikają z niedokrwienia i zwłóknienia wątroby. Funkcja wątroby z reguły nie cierpi, ale u 30-50% pacjentów obserwuje się niewielki wzrost aktywności aminotransferaz w surowicy, zwykle nie więcej niż 2 razy, czasami 7-8 razy.
Zespół cytopeniczny - Najwcześniejszym i najbardziej charakterystycznym objawem jest małopłytkowość z samoistnym zespołem krwotocznym w postaci krwiaków podskórnych, krwawień z błon śluzowych lub przedłużającego się krwawienia po drobnych zabiegach chirurgicznych. Następnie rozwija się niedokrwistość i leukopenia ze względną limfocytozą i bezwzględną neutropenią, ale nie obserwuje się wyraźnego wzrostu częstości występowania chorób zakaźnych u pacjentów.
Uszkodzenie kości waha się od bezobjawowej osteopenii i deformacji dystalnych kości udowych w kształcie kolby (kolba Erlenmeyera) do ciężkiej osteoporozy i martwicy niedokrwiennej (jałowej) z rozwojem wtórnej choroby zwyrodnieniowej stawów. Uszkodzenia układu kostno-stawowego mogą objawiać się ostrym lub przewlekłym bólem, złamaniami patologicznymi oraz rozwojem nieodwracalnych wad ortopedycznych wymagających leczenia operacyjnego (wymiana stawu). U dzieci i młodych dorosłych charakterystyczny jest rozwój tzw. kryzysów kostnych – epizodów silnego bólu, któremu towarzyszy gorączka i miejscowe ostre objawy zapalne (obrzęk, zaczerwienienie), symulujące obraz zapalenia kości i szpiku. Czynnikiem ryzyka rozwoju przełomów kostnych i poważnych uszkodzeń układu kostno-stawowego jest splenektomia, która predysponuje do rozwoju zespołu nadkrzepliwości i niedokrwiennego uszkodzenia kości (martwicy kości), będącej przyczyną przełomów kostnych. Uszkodzenie układu kostno-stawowego jest z reguły głównym problemem klinicznym w chorobie Gauchera typu I, decydującym o ciężkości choroby i jakości życia chorych.
Objawy uszkodzenia ośrodkowego układu nerwowego występują jedynie w neuropatycznych typach choroby Gauchera u dzieci (typ II i III) i mogą obejmować apraksję okoruchową lub zez zbieżny, ataksję, zaburzenia czucia i postępującą utratę inteligencji.
Uszkodzenie płuc występuje u 1-2% chorych, głównie u tych, którzy przeszli splenektomię i objawia się śródmiąższowymi zmianami w płucach lub uszkodzeniem naczyń płucnych z rozwojem objawów nadciśnienia płucnego.
2. Diagnostyka
2.1 Skargi i wywiad
przebyta splenektomia (całkowita lub częściowa);
ból kości i stawów; czas trwania, charakter i lokalizacja bólu, obecność złamań kości w przeszłości; objawy spontanicznego zespołu krwotocznego lub powikłań krwotocznych podczas interwencji chirurgicznych;
dolegliwości anemiczne, objawy stanu hipermetabolicznego (niewielka gorączka, utrata masy ciała);
obciążony wywiad rodzinny (obecność splenektomii lub powyższych objawów u rodzeństwa).
2.2 Badanie fizykalne
Zalecana przeprowadzić badanie, w tym zmierzyć wzrost i masę ciała, temperaturę ciała; ocena stanu układu kostno-stawowego; identyfikacja oznak zespołu krwotocznego; obecność hepatosplenomegalii, limfadenopatii; obecność oznak dysfunkcji serca, płuc, wątroby i narządów układu hormonalnego.
2.3 Diagnostyka laboratoryjna
Enzymodiagnostyka - wykrywanie aktywności kwaśna a-glukocerebrozydaza w leukocytach krwi lub w hodowanych fibroblastach uzyskanych z biopsji skóry.
Uwagi: rozpoznanie zostaje potwierdzone, gdy aktywność enzymu spadnie do poziomu poniżej 30% wartości prawidłowej (kategoria A). Stopień obniżenia aktywności enzymu nie koreluje z nasileniem objawów klinicznych i przebiegiem choroby.
Analiza molekularna mutacji genu glukocerebrozydazy.
Uwagi: analiza molekularna w celu wykrycia mutacji w genie glukocerebrozydazy pozwala na weryfikację rozpoznania choroby Gauchera, ale nie jest metodą diagnostyczną obowiązkową i jest wykorzystywana w diagnostyce różnicowej w skomplikowanych przypadkach klinicznych lub do analiz naukowych.
Analiza morfologiczna szpiku kostnego (nakłucie mostka i/lub biopsja trepanacyjna szpiku): u dorosłych pacjentów należy koniecznie wykluczyć inną przyczynę hepatosplenomegalii, do której zalicza się hemoblastozy i nienowotworowe choroby układu krwionośnego. U dzieci badanie szpiku kostnego wykonuje się wyłącznie w szczególnych wskazaniach.
Uwagi:badanie morfologiczne szpiku kostnego wykazuje charakterystyczne objawy diagnostyczne – liczne komórki Gauchera. Sporadycznie pojedyncze komórki o podobnej morfologii (komórki pseudo-Gauchera) stwierdza się w innych chorobach, którym towarzyszy zwiększone niszczenie komórek, np. w przewlekłej białaczce szpikowej i chorobach limfoproliferacyjnych i są wyrazem przeciążenia układu makrofagów produktami degradacji komórek klon białaczkowy.
Analiza kliniczna krwi i moczu
Chemia krwi, w tym:
rutynowe wskaźniki: bilirubina całkowita i bezpośrednia; aktywność aminotransferaz, fosfatazy alkalicznej, transpeptydazy β-glutamylowej, dehydrogenazy mleczanowej; mocznik, kreatynina, cholesterol, trójglicerydy, glukoza, białko całkowite, albuminy, elektroforeza globulin;
zastępcze markery aktywności choroby Gauchera (chitotriozydaza i/lub chemokina w surowicy CCL-18);
wskaźniki metabolizmu żelaza w surowicy (żelazo, całkowita zdolność wiązania żelaza w surowicy, ferrytyna, transferyna);
stężenie witaminy B12 i kwasu foliowego w surowicy (u dorosłych).
Badanie koagulogramu(aPTT, protrombina, fibrynogen, agregacja płytek krwi)
Oznaczanie markerów w surowicy wirusa zapalenia wątroby typu B i C(HBsAg i anty-HCV)
Badanie immunochemiczne białek surowicy z oznaczaniem immunoglobulin klas G, A, M, paraprotein, krioglobulin
2.4 Diagnostyka instrumentalna
Określenie ciężkości choroby Gauchera i ewentualnych chorób współistniejących Zalecana wykonanie następujących badań:
USG narządów jamy brzusznej i nerek
RTG kości udowych (w tym stawów kolanowych i biodrowych)
MRI kości udowej
Badanie MRI lub CT wątroby i śledziony z określeniem objętości narządów (cm3)
rejestracja elektrokardiogramu
Uwagi: USG i TK wątroby i śledziony pozwalają na identyfikację ich zmian ogniskowych i określenie początkowej objętości narządów, co jest niezbędne do późniejszego monitorowania skuteczności ERT.
2.5 Konsultacje specjalistyczne
Ortopeda;
Neurolog (wg wskazań)
Ginekolog (wg wskazań)
Okulista (wg wskazań)
Kardiolog (wg wskazań)
2.6 Badania dodatkowe
Echokardiografia dopplerowska – u pacjentów po splenektomii
Ezofagogastroduodenoskopia - w przypadku niestrawności lub objawów nadciśnienia wrotnego
RTG pozostałych części układu kostno-stawowego w przypadku występowania dolegliwości bólowych lub schorzeń narządu ruchu w tych częściach
Densytometria kości szkieletowych (standardowo - kręgi lędźwiowe i szyjka kości udowej).
Uwagi: Densytometria kości szkieletowych jest badaniem obowiązkowym w przypadku przebytych patologicznych złamań kości (standardowo – kręgosłupa lędźwiowego i szyjki kości udowej).
Siła rekomendacji: B (poziom wiarygodności: 2)3. Leczenie
3.1 Enzymatyczna terapia zastępcza
Choroba Gauchera jest pierwszą dziedziczną enzymopatią, dla której opracowano wysoce skuteczną enzymatyczną terapię zastępczą (ERT). Do chwili obecnej światowe doświadczenie w leczeniu choroby Gauchera rekombinowaną glukocerebrozydazą wynosi około 20 lat i stanowi „złoty standard” w leczeniu tej choroby. Jednak ze względu na niewielką liczbę pacjentów na świecie skuteczność ERT opiera się wyłącznie na ocenie obserwacji klinicznych, gdyż ze względów etycznych nie przeprowadzono specjalnych randomizowanych badań kontrolowanych placebo. Siła rekomendacji: C (poziom wiarygodności: 3). W Federacji Rosyjskiej ERT jest świadczona pacjentom z chorobą Gauchera w ramach państwowego programu „7 nozologii” od 2007 roku.
Do głównych celów leczenia pacjentów z chorobą Gauchera zalicza się:
eliminowanie bólu, normalizowanie samopoczucia pacjentów
regresja lub osłabienie zespołu cytopenicznego
zmniejszenie wielkości śledziony i wątroby
zapobieganie nieodwracalnym uszkodzeniom układu mięśniowo-szkieletowego i ważnych narządów wewnętrznych (wątroba, płuca, nerki).
3.2 Leczenie zachowawcze
dzieciństwo,
cytopenia,
kliniczne i radiologiczne objawy uszkodzenia kości (z wyjątkiem łagodnej osteopenii i kolbowatej deformacji dystalnych kości udowych – „kolby Erlenmeyera”),
znaczne powiększenie śledziony i wątroby,
znaczne powiększenie wątroby u pacjentów po splenektomii,
objawy uszkodzenia płuc i innych narządów.
Wskazania do rozpoczęcia enzymatycznej terapii zastępczej:
W Federacji Rosyjskiej zarejestrowane są 2 leki rekombinowane zawierające glukocerebrozydazę:
Imigluceraza syntetyzowana przez linię komórkową uzyskaną z jajników chomika chińskiego;
Welagluceraza alfa jest wytwarzana przez linię komórkową ludzkich fibroblastów HT-1080.
Uwagi:imiglucerazę i welaglucerazę podaje się dożylnie raz na 2 tygodnie. Formą uwalniania tych leków są butelki po 400 jednostek. Zawartość każdej fiolki (imigluceraza, welagluceraza) rozpuszcza się w wodzie do wstrzykiwań i ostrożnie miesza, unikając tworzenia się pęcherzyków. Cały przygotowany roztwór zbiera się do jednej butelki i rozcieńcza 0,9% roztworem chlorku sodu do wstrzykiwań dożylnych do całkowitej objętości 150-200 ml. Lek podaje się dożylnie w ciągu 1-2 godzin. Leku nie należy podawać jednocześnie z innymi lekami. Leczenie charakteryzuje się doskonałą tolerancją i wysoką skutecznością kliniczną u pacjentów z chorobą Gauchera typu 1.
Dawka początkowa rekombinowanej glukocerebrozydazy jest przedmiotem debaty i w różnych krajach waha się od 10 do 60 j./kg masy ciała przy częstotliwości podawania co 2 tygodnie. Przy ustalaniu dawki bierze się pod uwagę wiek pacjenta, charakter i nasilenie objawów klinicznych, rokowanie przebiegu choroby, obecność powikłań i chorób współistniejących. W krajach, które zapewniają bezpłatną ERT w ramach programu rządowego, istnieją rady ekspertów ds. choroby Gauchera, których zadaniem jest przepisywanie i monitorowanie skuteczności ERT.
W Federacji Rosyjskiej u dorosłych pacjentów z ciężkimi objawami choroby Gauchera typu I dawka początkowa imiglucerazy/welaglucerazy wynosi 30 j. na kg masy ciała, w postaci kroplówki dożylnej 2 razy w miesiącu.
Uwagi: W niektórych przypadkach (ciężka osteoporoza z powtarzającymi się patologicznymi złamaniami kości cewkowych; uszkodzenie płuc z rozwojem nadciśnienia płucnego lub zespołu wątrobowo-płucnego) dawkę rekombinowanej glukocerebrozydazy można zwiększyć do 60 j./kg na jedno podanie, ale decyzja w tym zakresie zostanie podjęta przez Radę Ekspertów utworzoną 01.04.2009 r. wsparcie Ministerstwa Zdrowia Federacji Rosyjskiej. Po osiągnięciu celów terapeutycznych u dorosłych pacjentów dawkę ERT zmniejsza się stopniowo do dawki podtrzymującej 7,5-15 j./kg 1-2 razy w miesiącu (przez całe życie). Schemat leczenia podtrzymującego jest w fazie opracowywania.
U dzieci z chorobą Gauchera dawka początkowa TRT wynosi:
W przypadku I i III typu choroby, która przebiega bez uszkodzenia rurkowatych kości szkieletu – 30 j./kg co 2 tygodnie;
W przypadku I i III typu choroby, która przebiega z uszkodzeniem rurkowatych kości szkieletu (przełomy kostne, złamania patologiczne, ogniska zniszczenia litycznego, aseptyczna martwica głów kości udowych) – 60 j.m./kg co 2 tygodnie.
Uwagi:Aby uzyskać maksymalny efekt w odniesieniu do wszystkich objawów klinicznych choroby, konieczne jest opracowanie zindywidualizowanego planu leczenia, który opiera się na fachowej ocenie ciężkości choroby Gauchera i obejmuje badanie pacjenta w specjalistycznej placówce medycznej ze specjalistami m.in. o różnych profilach, którzy mają duże doświadczenie w diagnozowaniu i leczeniu tej choroby. W Federacji Rosyjskiej badanie dorosłych pacjentów w celu oceny ciężkości choroby Gauchera i ustalenia dawki początkowej ERT przeprowadza się w Centrum Gauchera na podstawie wydziału naukowo-klinicznego chorób sierocych Federalnej Państwowej Instytucji Budżetowej „Hematologiczne Centrum Badawcze” Ministerstwa Zdrowia Federacji Rosyjskiej; podstawowe badanie dzieci - w Federalnej Państwowej Instytucji Budżetowej „Naukowe Centrum Zdrowia Dzieci” lub Federalnej Państwowej Instytucji Budżetowej „Federalne Centrum Naukowo-Kliniczne Hematologii, Onkologii i Immunologii Dziecięcej im. Dmitrija Rogaczowa” Ministerstwa Zdrowia Rosji Federacja.
3.3 Leczenie ortopedyczne
Wskazania do operacyjnego leczenia ortopedycznego ustalają chirurdzy ortopedzi mający doświadczenie w monitorowaniu i leczeniu pacjentów z chorobą Gauchera, przy udziale hematologów, radiologów i w razie potrzeby innych specjalistów zajmujących się leczeniem tego pacjenta. Wskazane jest przeprowadzanie planowych operacji ortopedycznych w placówkach medycznych specjalizujących się w diagnostyce i leczeniu chorób sierocych, które posiadają doświadczenie w leczeniu operacyjnym pacjentów z chorobą Gauchera i możliwość terapii zastępczej składnikami krwi w przypadku powikłań krwotocznych ( dla dorosłych pacjentów – oddział chorób sierocych Państwowego Centrum Naukowego Ministerstwa Zdrowia Federacji Rosyjskiej).
4. Rehabilitacja
Pacjentom z uszkodzeniami układu kostno-stawowego i/lub po endoprotezoplastyce stawu zaleca się rehabilitację w sanatoriach ortopedycznych, terapię ruchową i kinezyterapię.
5. Profilaktyka i obserwacja kliniczna
Nie ma możliwości zapobiegania chorobie Gauchera jako dziedzicznej chorobie metabolicznej.
2) diagnostyka prenatalna choroby Gauchera typu 2 i 3 w celu szybkiego podjęcia decyzji w sprawie przerwania ciąży u kobiet, które wcześniej rodziły dzieci z chorobą Gauchera typu 2-3.
5.1 Monitorowanie przebiegu choroby Gauchera i ocena skuteczności ERT
Dynamiczne monitorowanie pacjentów z chorobą Gauchera obejmuje badania okresowe oraz badania laboratoryjne (ogólne i biochemiczne badania krwi), których częstotliwość zależy od wieku chorych, czasu trwania i fazy ERT (tab. 3 i 4).
W celu oceny skuteczności leczenia i dostosowania dawki rekombinowanej glukocerebrozydazy raz na 1-3 lata przeprowadza się badanie kontrolne pacjentów z oceną wyników badań wykonywanych przez specjalistów o różnych profilach: terapeuta-hematolog, radiolog, ortopeda, neurolog, kardiolog z doświadczeniem w diagnostyce i leczeniu choroby Gauchera. Badanie kontrolne obejmuje ogólne badanie terapeutyczne (patrz wyżej), badania laboratoryjne i instrumentalne oraz konsultacje specjalistów.
Tabela 3– Schemat monitorowania dorosłych pacjentów z chorobą Gauchera
|
Pacjenci nieotrzymujący TRT |
Pacjenci otrzymujący TRT |
|||||
|
Cele leczenia nie zostały osiągnięte |
Osiągnięte cele leczenia |
|||||
|
Co 12 miesięcy |
12-24 miesiące . |
Co 3-6 miesięcy. |
Co 12 miesięcy |
|||
|
Analiza krwi |
||||||
|
Biochemia |
||||||
|
Metabolizm żelaza + kwasu foliowego + witaminy B12 |
||||||
|
Objętość śledziony (MRI lub CT) |
||||||
|
Objętość wątroby |
||||||
|
MRI kości udowej |
||||||
|
Rentgen kości |
5.2 Cechy monitorowania choroby Gauchera u dzieci
Monitorowanie przebiegu choroby Gauchera u dzieci odbywa się zgodnie z zaleceniami opracowanymi przez Joint International Group for the Study of Gauchera Disease. Badanie kliniczne przez pediatrę przeprowadza się co 2 tygodnie przed podaniem leku. Schemat monitorowania dzieci z chorobą Gauchera na tle ERT przedstawiono w tabeli. 4.
Tabela 4 – Schemat monitorowania dzieci z chorobą Gauchera
|
Pacjenci nieotrzymujący TRT |
Pacjenci otrzymujący TRT |
|||||||
|
Pierwszy rok obserwacji |
Po roku obserwacji |
W okresie zmiany dawki lub rozwoju powikłań klinicznych |
||||||
|
Co 12 miesięcy |
Każdego miesiąca |
Co 3-4 miesiące |
Co 12 miesięcy |
Co 3-4 miesiące. |
Co 6 miesięcy |
Co 12 miesięcy |
||
|
Badanie pediatry |
||||||||
|
Analiza krwi |
||||||||
|
Biochemia |
||||||||
|
Biomarkery (chitotriozydaza) |
||||||||
|
Metabolizm żelaza |
||||||||
|
Objętość śledziony (MRI lub CT) |
||||||||
|
Objętość wątroby |
||||||||
|
Rentgen kości |
||||||||
|
Badanie gęstości kości |
6. Informacje dodatkowe mające wpływ na przebieg i wynik choroby
6.1 Prognoza
W chorobie Gauchera typu I rokowanie jest korzystne, jeśli ERT zostanie przepisana w odpowiednim czasie. Wraz z rozwojem nieodwracalnych uszkodzeń układu kostno-stawowego wskazane jest chirurgiczne leczenie ortopedyczne w celu korekcji wad ortopedycznych. W przypadku uszkodzenia ważnych narządów wewnętrznych rokowanie zależy od stopnia dysfunkcji dotkniętych narządów i rozwoju powikłań (na przykład krwawienie z żylaków przełyku i żołądka u pacjentów z marskością wątroby i nadciśnieniem wrotnym; niepowodzenia u pacjentów z uszkodzeniem płuc).
6.2 Błędy i nieuzasadnione przypisania
- Splenektomia nie jest zalecana.
Uwagi: Jeśli zostanie ustalona diagnoza choroby Gauchera, splenektomia jest możliwa tylko w przypadku bezwzględnych wskazań (na przykład urazowe pęknięcie śledziony). Jeżeli u osób z niejasną splenomegalią i cytopenią konieczna jest splenektomia, zaleca się wykluczenie rozpoznania choroby Gauchera.
- Powtarzane nakłucia szpiku kostnego i inne inwazyjne badania diagnostyczne (biopsja wątroby, śledziona) przy potwierdzonym rozpoznaniu choroby Gauchera nie są konieczne.
- chirurgiczne leczenie przełomów kostnych, które są błędnie uważane za objawy zapalenia kości i szpiku
- przepisanie glikokortykosteroidów w celu złagodzenia zespołu cytopenicznego
- przepisywanie suplementów żelaza nieleczonym pacjentom z chorobą Gauchera, ponieważ niedokrwistość w tych przypadkach ma charakter „niedokrwistości zapalnej”.
6.3 Choroba Gauchera i ciąża
Choroba Gauchera nie jest przeciwwskazaniem do zajścia w ciążę. Wskazane jest planowanie ciąży po osiągnięciu celów leczenia choroby Gauchera. Kwestia kontynuacji ERT w czasie ciąży i karmienia piersią jest ustalana indywidualnie, biorąc pod uwagę stan pacjentki i jej przestrzeganie leczenia. Prowadzenie ciąży prowadzą doświadczeni położnicy-ginekolodzy wspólnie z hematologiem. O sposobie porodu decydują wskazania położnicze, biorąc pod uwagę obecność cytopenii i stan układu hemostatycznego.
Kryteria oceny jakości opieki medycznej
|
Kryteria jakości |
Ocena wydajności |
Poziom dowodów |
||
|
Przy postawieniu diagnozy wykonywano oznaczenie aktywności β-D-glukozydazy w leukocytach krwi obwodowej, zaschniętych plamach krwi i/lub badania genetyki molekularnej (wykrywanie mutacji w genie GBA kodującym β-D-glukozydazę). |
||||
|
Wykonano kliniczne badanie krwi (płytki krwi, erytrocyty, hemoglobina, leukocyty) |
||||
|
Wielkość wątroby i śledziony określano za pomocą USG lub MRI narządów jamy brzusznej |
||||
|
W przypadku patologii kości konieczna była konsultacja ortopedyczna |
||||
|
Wykonano zdjęcia RTG kości udowych, jeśli nie były wykonywane w ciągu ostatnich 12-24 miesięcy |
||||
|
Wykonano biochemiczne badanie krwi (białko całkowite, kreatynina, ALT, AST, bilirubina całkowita i bezpośrednia, cholesterol, trójglicerydy, LDH), jeśli nie wykonano go w ciągu ostatnich 12 miesięcy |
Bibliografia
Przewodnik po hematologii // Pod. wyd. sztuczna inteligencja Worobiow. – W 3 tomach – M.: Newdiamed. – 2003. – T. 2 – s. 202-205.
Krasnopolska K.D. Dziedziczne choroby metaboliczne // M.: 2005. – s. 20-22.
Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. Gen i pseudogen ludzkiej glukocerebrozydazy: struktura i ewolucja // Genomika 1989 styczeń; 4(1): 87-96.
Choroba Gauchera / wyd. A.H. Futermana i A. Zimrana. – Taylor & Francis Group, LLC, 2007. – 528 s.
Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, Grabowski GA, Mistry PK, Tylki-Szymańska A. Cele terapeutyczne w leczeniu choroby Gauchera. Nasienie Hematol. Październik 2004; 41 (4 Suppl 5): 4-14.
Mistry PK i Cox TM. Locus glukocerebrozydazy w chorobie Gauchera: analiza molekularna enzymu lizosomalnego // J Med Genet. 1993 listopad; 30(11): 889–894.
Grabowski G.A. Choroba Gauchera i inne zaburzenia przechowywania // W: Hematologia 2012: 54. Doroczne Spotkanie i Wystawa ASH. Atlanta, Gruzja, 2012; 13-18.
Boven LA, van Meurs M, Boot RG i in. Komórki Gauchera wykazują odrębny fenotyp makrofagów i przypominają alternatywnie aktywowane makrofagi // Am J Clin Pathol. 2004;122:359-369.
Mikosz P. Artykuł od redakcji: Choroba Gauchera // Wien Med Wochenschr. Grudzień 2010; 160(23-24):593.
Mankin HJ, Rosenthal DI, Xavier R. Choroba Gauchera. Nowe podejście do starożytnej choroby // J Bone Joint Surg Am. maj 2001; 83-A(5):748-62.
Lukina E.A. Choroba Gauchera // M.: Literra. – 2011. – 54 s.
Lukina K.A. Czynniki kliniczne i molekularne związane z uszkodzeniem układu kostno-stawowego w chorobie Gauchera typu I: dis. ...cad. Miód. Nauka. - Moskwa. – 2013. – 142 s.
Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, Beutler E. Choroba Gauchera. Cechy kliniczne, laboratoryjne, radiologiczne i genetyczne 53 pacjentów. Medycyna (Baltimore). 1992 listopad;71(6):337-53.
Stein P, Yu H, Jain D, Mistry PK. Hiperferrytynemia i przeciążenie żelazem w chorobie Gauchera typu 1. Jestem J Hematol. 2010;85(7):472-476.
Wenstrup RJ, Roca-Espiau M, Weinreb NJ i in., Szkieletowe aspekty choroby Gauchera: przegląd // Br J Radiol, 2002; 75 (dodatek 1): A2 – A12.
CoxTM, Schofield JP. Choroba Gauchera: cechy kliniczne i historia naturalna // Baillieres Clin Haematol. 1997;10:657-89.
Kaplan P., Baris H., De Meirleir L. i in. Zmienione zalecenia dotyczące postępowania w chorobie Gauchera u dzieci // Eur J Pediatr. kwiecień 2013;172(4):447-58.
Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C, Weinreb NJ, Cox TM. Terapia zastępcza enzymami i redukcja substratu w chorobie Gauchera. System bazy danych Cochrane Rev. 27 marca 2015 r.;(3):CD010324. doi: 10.1002/14651858.CD010324.pub2.
Mistry P.K., Cappellini M.D., Lukina E. i in. Konferencja konsensusowa: Ponowna ocena choroby Gauchera – algorytmy diagnostyki i leczenia choroby. Jestem J Hematol. styczeń 2011; 86 ust. 1: 110–115
Dodatek A1. Skład grupy roboczej
Lukina E.A.1, doktor nauk medycznych, profesor, kierownik. Zakład Naukowo-Kliniczny Chorób Sierocych
Sysoeva E.P.1, dr, starszy pracownik naukowy
Mamonov V.E.1, dr, kierownik. Zakład Ortopedii Hematologicznej
Yatsyk G.A.1, dr, kierownik. oddział rezonansu magnetycznego i USG
Doktor Tsvetaeva N.V.1, wiodący badacz
Gundobina O.S.2, dr, kierownik. szpital dzienny
Savostyanov K.V. 2, dr hab., kierownik. Pracownia Genetyki Molekularnej i Biologii Komórki
Wiszniewa E.A. 2, doktor nauk medycznych, zastępca dyrektorzy
Finogenova N.A. 3, doktor nauk medycznych, profesor, starszy pracownik naukowy
Smetanina N.S. 3, doktor nauk medycznych profesor, zastępca dyrektorzy
Federalna Państwowa Instytucja Budżetowa Hematologiczne Centrum Naukowe Ministerstwa Zdrowia Federacji Rosyjskiej, Moskwa
Federalna Państwowa Instytucja Budżetowa „Centrum Naukowe Zdrowia Dzieci” Ministerstwa Zdrowia Federacji Rosyjskiej, Moskwa
Federalna Państwowa Instytucja Budżetowa Federalne Centrum Badań Naukowych w zakresie Hematologii, Onkologii i Immunologii Dziecięcej im. D. Rogaczowa Ministerstwo Zdrowia Federacji Rosyjskiej, Moskwa
Projekt wytycznych klinicznych został zweryfikowany 29 października 2013 r. na posiedzeniu Grupy Ekspertów ds. Chorób Rzadkich Departamentu Chorób Sierocych Federalnej Państwowej Instytucji Budżetowej Państwowego Centrum Naukowego Ministerstwa Zdrowia Federacji Rosyjskiej, na posiedzeniu Komisji Profilowej ds. specjalności „Hematologia” w lutym 7 listopada 2014 r., na posiedzeniu Rady Ekspertów ds. Chorób Sierocych w dniu 24 września 2014 r., zatwierdzony na posiedzeniu Komisji Profilowej dla specjalności „Hematologia” 7 listopada 2014 r.
Specjaliści hematolodzy;
Specjaliści pediatryczni
Specjaliści terapeuci;
Specjaliści gastroenterolodzy/hepatolodzy;
Specjaliści chorób zakaźnych;
Specjaliści ortopedzi
Studenci medycyny
Metodologia gromadzenia dowodów
Metody stosowane w celu gromadzenia/wyboru materiału dowodowego:
Poszukaj publikacji w czasopismach specjalistycznych o współczynniku udarności > 0,3;
Szukaj w elektronicznych bazach danych.
Bazy danych wykorzystywane do gromadzenia/wyboru materiału dowodowego:
Metody stosowane do analizy dowodów:
Przeglądy systematyczne z tabelami dowodowymi.
Metody stosowane w celu sprawdzenia jakości i siły dowodów:
Konsensus ekspertów;
Ocena znaczenia dowodów zgodnie ze schematem oceny dowodów (Tabela A1).
|
Poziomy dowodów |
Opis |
|
Wysokiej jakości metaanalizy, systematyczne przeglądy randomizowanych badań kontrolowanych (RCT) lub RCT o bardzo niskim ryzyku błędu systematycznego |
|
|
Wysokiej jakości metaanalizy, przeglądy systematyczne lub RCT |
|
|
Metaanalizy, przeglądy systematyczne lub RCT obarczone wysokim ryzykiem błędu systematycznego |
|
|
Wysokiej jakości przeglądy systematyczne badań kliniczno-kontrolnych lub badań kohortowych, w których nie występuje ryzyko wystąpienia zakłócających skutków lub stronniczości lub jest ono bardzo niskie, a prawdopodobieństwo wystąpienia związku przyczynowego jest wysokie |
|
|
Dobrze przeprowadzone badania kliniczno-kontrolne lub badania kohortowe z umiarkowanym ryzykiem efektów zakłócających lub stronniczości oraz umiarkowanym prawdopodobieństwem związku przyczynowego |
|
|
Badania kliniczno-kontrolne lub badania kohortowe z wysokim ryzykiem efektów zakłócających lub stronniczości oraz umiarkowanym prawdopodobieństwem związku przyczynowego |
|
|
Badania nieanalityczne (opisy przypadków, serie przypadków) |
|
|
Opinia eksperta |
Opis metodologii analizy dowodów i opracowywania rekomendacji
Wybierając publikacje jako potencjalne źródła dowodów, sprawdzono metodologię zastosowaną w każdym badaniu, aby upewnić się, że jest ona zgodna z zasadami medycyny opartej na faktach. Wynik badania wpłynął na poziom materiału dowodowego przypisanego publikacji, co z kolei wpłynęło na siłę wynikających z niej rekomendacji.
Badanie metodologiczne skupiało się na cechach projektu badania, które miały istotny wpływ na jakość wyników i wniosków.
Aby wykluczyć wpływ czynników subiektywnych, każde badanie było oceniane niezależnie przez co najmniej dwóch niezależnych członków zespołu autorskiego. Różnice w ocenach omawiano na spotkaniach grupy roboczej autorów tych rekomendacji.
Na podstawie analizy dowodów konsekwentnie opracowano fragmenty zaleceń klinicznych z oceną siły zgodnie ze schematem oceny zaleceń (tab. A2).
Metody stosowane przy formułowaniu rekomendacji:
Konsensus ekspertów;
Wewnętrzna ocena ekspercka;
Ocena eksperta zewnętrznego.
Poziomy dowodów.
Poziom A. Dowody opierają się na danych z wielu randomizowanych badań klinicznych lub metaanaliz, przeglądów systematycznych.
Poziom B: Dowody oparte na danych z jednego randomizowanego badania klinicznego lub wielu badań nierandomizowanych.
Poziom C. Konsensus ekspertów i/lub ograniczone badania, badania retrospektywne, rejestry.
Poziom D. Opinia biegłego.
Tabela P2 Wiarygodność dowodów
Punkty dobrych praktyk (GPP):
Ocena eksperta zewnętrznego;
Wewnętrzna ocena ekspercka.
Projekty wytycznych zostały zweryfikowane przez niezależnych ekspertów, których poproszono o skomentowanie jakości interpretacji dowodów i opracowania zaleceń. Dokonano także eksperckiej oceny sposobu prezentacji rekomendacji i ich przystępności do zrozumienia.
Wersja ostateczna:
W celu ostatecznej weryfikacji i kontroli jakości rekomendacje zostały ponownie przeanalizowane przez członków zespołu autorskiego, którzy doszli do wniosku, że uwzględniono wszystkie istotne uwagi i uwagi ekspertów, a ryzyko błędów systematycznych w opracowaniu zostało zminimalizowane.
Najnowsze zmiany i ostateczna wersja tych zaleceń zostały sprawdzone i zatwierdzone w dniu 24 września 2014 r. na posiedzeniu Wielodyscyplinarnej Rady Ekspertów ds. Chorób Sierocych w Centrach Federalnych Ministerstwa Zdrowia Federacji Rosyjskiej.
Dodatek B. Algorytmy postępowania z pacjentem
Algorytm rozpoznawania choroby Gauchera i postępowania z pacjentami
Załącznik B: Informacje dla pacjenta
W sercu choroby Gauchera występuje dziedziczny niedobór aktywności enzymu β-glukocerebrozydazy, który bierze udział w przetwarzaniu produktów metabolizmu komórkowego (metabolizm). W wyniku niedostatecznej aktywności tego enzymu nieprzetworzone „odpady” metaboliczne gromadzą się w komórkach „zmiataczy” (makrofagach), a komórki te przyjmują charakterystyczny wygląd komórek Gauchera, czyli „komórek magazynujących”. Komórki przepełnione „odpadami produkcyjnymi” gromadzą się niczym w magazynie w narządach wewnętrznych, najpierw w śledzionie, następnie w wątrobie, kościach szkieletowych, szpiku kostnym i płucach (stąd określenie „choroba spichrzowa”). Choroba Gauchera występuje z częstością od 1:40 000 do 1:60 000 we wszystkich grupach etnicznych; w populacji Żydów aszkenazyjskich częstość występowania tej choroby sięga 1:450.
Główne objawy choroby Gauchera spowodowane są nagromadzeniem komórek przeciążonych „odpadami” i dysfunkcją tych komórek. Nagromadzenie komórek w różnych narządach prowadzi do zwiększenia ich rozmiaru (śledziona, wątroba) i/lub zaburzenia struktury i funkcji (kości, szpik kostny, płuca). Zakłócenie funkcjonowania komórek (makrofagów), przeciążonych odpadami, skutkuje rozwojem anemii, krwawień, wyczerpania, łamliwości kości i przełomów bólowych. Wynika to z faktu, że zakres „obowiązków zawodowych” makrofagów w organizmie człowieka jest bardzo szeroki i obejmuje regulację wielu procesów życiowych: hematopoezy, krzepnięcia krwi, obrotu kostnego itp. Najbardziej typowymi objawami choroby Gauchera są: zwiększenie wielkości śledziony i wątroby, rozwój niedokrwistości, trombocytopenii, przewlekłego bólu kości lub wystąpienia nagłych napadów silnego bólu kości (przełomów kostnych). Tym ostatnim towarzyszy gorączka i miejscowe ostre objawy zapalne (obrzęk, zaczerwienienie), przypominające obraz zapalenia kości i szpiku. Rzadziej choroba może najpierw objawiać się złamaniem kości w wyniku drobnego urazu. Zajęcie kości często stanowi główny problem kliniczny i może prowadzić do ciężkiej niepełnosprawności (bezruch na skutek licznych złamań patologicznych, deformacje kości i stawów, konieczność wymiany uszkodzonych stawów biodrowych lub barkowych).
Diagnostyka choroby Gauchera ustala się na podstawie analizy biochemicznej aktywności markera aktywności β-glukocerebrozydazy w leukocytach krwi. Spadek poziomu enzymu o mniej niż 30% normalnego poziomu potwierdza diagnozę.
Chorobę Gauchera można również zdiagnozować za pomocą analizy molekularnej genu glukocerebrozydazy.
Podobne artykuły
-
Marzyłam o welonie ślubnym
Dlaczego kobieta marzy o welonie: Dobrze znany symbol czystości, młodości, czystości, niewinności Widząc welon we śnie - taki sen obiecuje spotkanie i znajomość z osobą, która zmieni Twoje poglądy na temat życia. Jeśli marzyłeś...
-
Dlaczego śnisz o trzymaniu języka w ustach? Interpretacja snów o wyjmowaniu go z ust
Usta we śnie są symbolem komunikacji, wyrażania siebie, wskaźnikiem myśli i uczuć danej osoby. Dokładna i szczegółowa analiza własnego snu, a także związku pomiędzy tym, co widziałeś, a wydarzeniami zachodzącymi w prawdziwym życiu oraz poszukiwanie odpowiedzi w...
-
DO GOTOWANIA – przepisy na każdy dzień!
Czosnek to roślina wieloletnia, którą ludzie uprawiali już tysiąc lat temu, kiedy to młode pędy sprowadzono ze wschodu nawet do najodleglejszych zakątków planety. Pomimo zmiennego klimatu i trudnych warunków czosnek okazał się wytrwały...
-
Opis stanowiska sekretarza głowy
Sekretarz nazywany jest wiernym asystentem i prawą ręką szefa i nie bez powodu, ponieważ do obowiązków sekretarza menedżera należy zapewnienie skutecznego zarządzania i działań administracyjnych. Sekretarka kierownika jest zajęta...
-
Magia liczb Co oznacza wieniec we śnie?
Po obejrzeniu fabuły w duszy śniącego pozostaje nieprzyjemny posmak, niepokojące myśli w głowie nie dają spokoju. Co o tym myślą ezoterycy i interpretatorzy książek snów? Sen należy interpretować, biorąc pod uwagę specyfikę rozwoju fabuły snu,...
-
Dlaczego marzysz o rzece według wymarzonej książki?
Książka marzeń Millera Jeśli śnisz o gładkiej, spokojnej tafli rzeki, oznacza to, że wkrótce będziesz cieszyć się najcudowniejszymi radościami, a Twoje samopoczucie zachwyci Cię kuszącymi możliwościami. Jeśli wody rzeki są mętne i niespokojne -...