Синдром на Fanconi при деца и възрастни. Синдром на Fanconi: симптоми, диагностика и методи на лечение Как да се лекува този проблем
Синдромът на Fanconi (пълно име - de Toni-Debre-Fanconi) е вродена патология, която се изразява в сериозна дисфункция на проксималните бъбречни тубули, а именно нарушение на вторичната абсорбция (абсорбция в кръвта) на вещества, филтрирани от бъбреците , което води до глюкозурия (повишена захар в урината), фосфатурия (нарушена фосфорна и калциева обмяна), аминоацидурия (повишена екскреция на аминокиселини в урината) и намаляване на концентрацията на бикарбонати, които регулират киселинността на кръвта.
Синдром на Де Тони-Дебре-Фанкони
Синдромът на Fanconi е много рядко заболяване, което се среща главно при деца и според медицинската статистика честотата му съответства на 1 болно бебе на 350 хиляди новородени от двата пола.
При възрастни се наблюдава изключително рядко, развивайки се на фона на придобити патологии. Код на патологията според МКБ-10: E72.O.
причини
Природата и причините за генетичния дефект на синдрома на Fanconi днес не са добре разбрани.
Предполага се, че патологията се основава или на дефекти в транспортните протеини на бъбречните тубули, или на генна мутация, която нарушава функцията на ензимите, които регулират реабсорбцията на глюкоза, аминокиселини и фосфор.
Има доказателства от изследвания за точкови дефекти в митохондриите, водещи до неправилно функциониране на тубулите на бъбреците.
Заболяването е свързано и с непоносимост към фруктоза, хронично отравяне с токсини (тежки метали, ифосфамид, аминогликозиди), дефицит на витамин D, амилоидоза, недостатъчност на редица клетъчни ензими (пируват карбоксилаза, фосфоенолпируват карбоксикиназа и други), тирозинемия, метахроматична левкодистрофия, галактоземия, цистиноза, гликогеноза.
Според други експерти синдромът на Fanconi може да бъде изолирана патология - а именно една от тежките форми на рахитоподобни патологии, които са наследствени.
Проучванията потвърждават, че при синдрома на Fanconi клетъчният енергиен метаболизъм с участието на АТФ (аденозинтрифосфат) и междуклетъчния транспорт в тубулите на основния елемент на бъбрека, нефрона, са нарушени.
Поради недостатъчност на ензимните функции настъпват загуби на глюкоза, фосфати, аминокиселини и тубулите на бъбреците изпитват липса на енергия. В същото време с урината се отделят важни вещества, което води до дистрофични промени в костната тъкан - рахит.
Тъй като медицината все още не е стигнала до недвусмислено заключение относно причините за синдрома на Faconi, това състояние се нарича и с други термини: „глюкофосфаминов диабет“, „идиопатичен бъбречен синдром на Fanconi“, „D-резистентен рахит“, „бъбречен нанизъм с D-резистентен рахит” , „Наследствен синдром на Фанкони”.
Форми и патогенеза
Има два вида синдром на Fanconi:
- наследствен (вроден, идиопатичен), който се отнася до първичната форма;
- придобита, която се счита за вторична форма на заболяването.
Специалистите свързват наследствения (генетичен) вид с Х-хромозомен дефект, който се унаследява по доминантен и рецесивен начин, така че генетичното прогнозиране на проявата му в бъдещото потомство не е лесна задача. Ако патологията принадлежи към вроден тип (първична форма), тогава тя се открива при дете в гръдния период до една година. Следователно първичната форма се нарича "бебе".
Степента на генна мутация също определя тежестта на самия синдром. И така, наследственият пълен синдром на Fanconi се проявява в присъствието на 3 основни биохимични дефекта, които включват глюкозурия, аминоацидурия, фосфатурия, непълна - с две от тях.
Обикновено генетично обусловената патология е придружена от други вродени заболявания: цистиноза, синдроми на Уилсън, Дент, Лоу, непоносимост към фруктоза, тирозинемия (неспособността на тялото ефективно да разгражда аминокиселината тирозин), галактоземия (дисфункция на превръщането на захарта в глюкоза). ), прекомерно натрупване на гликоген.
Развитие на синдрома на Fanconi
Придобитият синдром (вторичен), за разлика от вродения, не е придружен, а е следствие от вече съществуващи патологии:
- тирозинемия тип I;
- цистиноза (нарушен метаболизъм на аминокиселината цистин с последващо увреждане на бъбреците);
- непоносимост към фруктоза;
- Болест на Уилсън-Коновалов;
- галактоземия;
- гликогеноза (анормално натрупване на гликоген в тъкани и органи) тип XI;
- наследствени патологии на бъбреците;
- (нарушение на протеиновия метаболизъм, което води до склероза, атрофия, органна дисфункция);
- тубулоинтерстициален (нефрит с увреждане на тъканите, бъбречни тубули);
- хиперпаратиреоидизъм (ендокринно заболяване, при което се нарушава нормалното съдържание на калций и фосфор в кръвта);
- злокачествени заболявания: миелом, бял дроб, панкреас, бял дроб, болест на леката верига;
- дълбоки изгаряния.
В допълнение, синдромът може да провокира такива състояния:
- трансплантация на органи с ниска тъканна съвместимост;
- дефицит на витамин D;
- отравяне с уран, бисмут, живак, олово, кадмий;
- контакт с толуен, малеинова киселина, лизол;
- употребата на нефротоксични фармакологични средства, като: гентамицин, платинови препарати, лекарства на основата на тетрациклин с изтекъл срок на годност, диданозин, цидофовир, противоракови химиотерапевтични лекарства - ифосфамид, стрептозоцин.
Симптоми и признаци
С наследствена (вродена) форма
Първичните симптоми се появяват през първите месеци от живота, рядко - след година и половина.
На първо място, при новородено дете се забелязват следните състояния:
- често уриниране (полиурия);
- повишена жажда (полидипсия);
- продължителен запек;
- чести пристъпи на безпричинно повръщане;
- астения (обща умора), мускулна слабост;
- необясними "скокове" на температурата до 37,5 - 38 C;
- подут корем.
Като правило, в периода, когато започват пристъпи на повръщане и температурата се повишава, бебето се показва на педиатър. Опитен специалист трябва да установи, че комбинацията от тревожни признаци за родителите не е свързана с остри респираторни инфекции, остри респираторни вирусни инфекции или ентеровирусна инфекция.
Компетентният педиатър е в състояние да разпознае синдрома на Fanconi навреме. И лабораторни изследвания - за потвърждаване на съмненията, ако разкрият три (или два) основни признака: глюкозурия, генерализирана хипераминоацидурия и хиперфосфатурия, характерни за тази патология.
След леки и доста неясни симптоми през следващата година и половина, симптомите, характерни за синдрома на Fanconi, са ясно фиксирани:
- Ранен нанизъм (нисък ръст), причинен от постоянната екскреция на най-важните аминокиселини, глюкоза, калций, фосфати от тялото. Първите шест месеца нормален растеж и тегло се заменят с дефицит на телесно тегло (до 30%) и растеж (от 2 до 21%).
- Рахитът, причинен от масивна екскреция на калций и фосфати, става забележим след 10-12 месеца от живота и има характеристики, характерни за синдрома на Fanconi: главата на бебето обикновено е леко деформирана, но големите кости на краката и ръцете показват значителна кривина - варусни деформации, когато пищялите на бебето са огънати от "колело" или валгус (под формата на буквата "X"). Костите на гръдния кош и гръбначния стълб също са огънати.
- Забавяне в умственото и физическото развитие.
- Липса на общителност, страх, комплекси.
- Полидипсията и полиурията могат да прогресират и регресират, без да изчезнат напълно.
- Умерена мускулна хипотония, изразяваща се в забавяне, затруднение на движенията, което води до факта, че децата на 5-6 години не могат да ходят.
- Болка в костите с умерена интензивност, пречеща на детето да ходи. Те са по-силно изразени на нивото на краката, таза и гръбначния стълб. Походката, ако детето ходи, става "патица", несигурна.
- Висока вероятност от фрактури на тръбни кости поради дефицит на минерали в костната тъкан.
- Остеомалация или омекване на костите поради разрушаване на костната тъкан в резултат на липса на калциеви и фосфорни соли.
- Намалена имунна защита срещу инфекции, което се проявява в чести вирусни заболявания, отит, пневмония.
- Парализа поради липса на калий.
- Офталмологични патологии, като пигментен ретинит, вродена катаракта.
- Развитие на патологии на нервната система, УНГ (ухо, нос, фаринкс, ларинкс) и стомашно-чревния тракт, сърдечно-съдовата система, анатомични аномалии на отделителната система поради масивни метаболитни нарушения.
- В единични случаи - ендокринни нарушения
С прогресирането на тубулните нарушения (нарушен транспорт на органични вещества, минерали, електролити) към 10-12-годишна възраст децата са по-склонни да развият хронична бъбречна недостатъчност, която застрашава живота им.
Видими симптоми на синдрома на Fanconi при деца 
При възрастни пациенти с развитие на вторична форма
Ако придобитият синдром на Fanconi се развие при възрастни с други заболявания или патологични състояния, тогава неговите прояви често се комбинират с проявите на провокаторната болест.
Основните характеристики обаче са:
- Увеличен обем на урината на ден (до 2 литра или повече) и остра жажда, които също са характерни за пациенти в ранна детска възраст.
- Обща и мускулна слабост, болки в костите.
- Висока вероятност от постоянно повишаване на кръвното налягане () на фона на бъбречна дисфункция.
- Остеомалация (разрушаване на костите).
- Ацидоза (повишена киселинност на кръвта), дължаща се на задържането на продукти на окисление в организма, което води до хипокалиемия (дефицит на калий).
- Нефрокалцинозата е високо отлагане на калциеви соли в бъбреците с висока температура, втрисане, гадене, силни болки в корема, слабините и яйчниците, които са характерни за това състояние.
- Хипокалиемия (нисък прием на калий), причиняваща тежки сърдечни усложнения, включително животозастрашаващи аритмии.
- Бързо (ако не се лекува) развитие на хронична бъбречна недостатъчност
Сред жените
Най-неблагоприятният курс на синдрома на Fanconi се проявява, когато се развива при жени в постменопауза. По това време, на фона на намаляване на производството на хормони, настъпва естествено намаляване на костната плътност (остеопения).
Когато това състояние се комбинира с повишена чупливост на костите поради липса на минерали, вероятността от тежки компресионни фрактури на прешлените, шийката на бедрената кост и последваща инвалидност е висока.
Диагностика
За потвърждаване или опровергаване на диагнозата се използват рентгенови лъчи за изследване на костите и задълбочени биохимични изследвания на кръвта и урината.
лаборатория
Откриваеми промени в биохимията на урината и кръвта:
| знаци | Индикатори |
|---|---|
| ниско съдържание на калций и фосфор | по-малко от 2,1 mmol/l и съответно 0,9 mmol/l |
| ацидоза ("подкисляване" на кръвта) | BE = 10 - 12 mmol / l |
| глюкозурия (повишена захар в урината) | 2-3% и повече |
| хипераминоцидурия (екскреция с урината на важни аминокиселини аланин, аргинин, глицин, отделяне) | до 2 - 2,5 g / ден |
| отделяне на калций в урината | 1,5 - 3,5 mmol / ден |
| повишаване на pH (киселинността) на урината поради необичайно висока загуба на бикарбонат | до 6.0 |
| повишаване на относителната плътност на урината | 1,025 – 1,035 |
Прегледът разкрива още:
- повишена активност на алкалната фосфатаза;
- прекомерно отделяне на натриеви и калиеви соли от тялото;
- протеинурия (появата на протеин в урината) в присъствието на леки вериги на имуноглобулини, лизозим, протеини с ниско молекулно тегло, бета 2-микроглобулини;
- повишен клирънс (скорост на филтриране) на пикочната киселина с нейното намалено серумно съдържание
- спад в активността на ензимите на енергийния метаболизъм: сукцинат дехидрогеназа, a-глицерофосфат дехидрогеназа, глутамат дехидрогеназа;
- повишаване на количеството на млечна и пирогроздена киселина в кръвта.
инструментална
Диагнозата на синдрома на Fanconi предвижда задължително използване на костна радиография, за да се открие деформация на скелета, крайниците, да се открият признаци на остеомалация, остеопороза, а при деца - допълнително - забавен растеж на костите в сравнение с нормата за календарната възраст.
Наблюдават се следните аномалии в костната тъкан:
- грубо влакнеста, клетъчна структура с празнини, слаба минерализация, необичайни израстъци под формата на "шипове" в бедрената кост и тибията;
- признаци на епифизиолиза (частично или пълно спиране на растежа на костите по дължина в незрелия скелет, което води до асиметрия на крайниците);
- фрактури на тръбни кости (на късен етап) на фона на развитието на остеопороза, чиято степен се определя с помощта на рентгенова денситометрия;
- натрупване на радиоизотоп в области на интензивен растеж на костите.
В бъбреците:
Електронно изследване на биопсия на бъбречна тъкан (биопсия) разкрива характерна промяна във формата на тубулите под формата на "лебедова шия", изтъняване, атрофия (намаляване на обема) на епителната тъкан в присъствието на увеличен брой митохондрии в него, фиброза (анормален растеж) на съединителната тъкан.
Необходими изследвания:
диференциал
Патологията трябва да се разграничава от всички изолирани заболявания, които могат да провокират появата на придобит синдром на Fanconi, придобити нездравословни състояния и интоксикации.
В допълнение, при кърмачета педиатърът е длъжен да разграничи състоянието на остър дефицит на витамин D при синдрома на Fanconi от неговия излишък при използване на изкуствени добавки или нарушения на калциевия метаболизъм.
Разлики в хипервитаминоза D и синдром на де Фанкони при деца под една година:
| Настроики | Хипервитаминоза D | синдром на де Фанкони |
|---|---|---|
| Честота | Често | Рядко |
| Симптоми (подобни) | Суха кожа, бледност, остра жажда, повръщане, продължителен запек, дефицит на тегло и височина, уголемяване на черния дроб | |
| Симптоми (различни) | Хипертония (чести покачвания на кръвното налягане) | Изтощение, полиурия, мускулна хипотония, липса на високо кръвно налягане |
| Кръв | Излишък на калций в острия период. Намалено съдържание на фосфор. Алкална фосфатаза, захар, протеин - в норма | Калцият често е нормален (може да бъде понижен). Глюкозата, протеинът са намалени. Фосфорът рязко намалява. Рязко се повишава активността на алкалната фосфатаза - 2-3 пъти. Повишена киселинност на кръвта |
| Урина | Тестът на Sulkovich за екскреция на калций е положителен. Наличие на протеин, кръв (повече от 2-3 еритроцита в зрителното поле), левкоцити. Захарният аминоазот обикновено е нормален | Пробата на Сулкович е отрицателна. Протеин, фосфати, повишена захар, аминоацидурия |
| Кости | Зоните на калцификация са разширени, уплътнени | Остеопороза на тубуларните кости, дефицит на калций в областите на калцификация |
Специалисти, занимаващи се със синдрома на Фанкони и неговите усложнения: нефролог, ортопед, хематолог, ендокринолог, уролог, офталмолог, генетик.
Лечение
Рационалната, добре разработена терапия може да намали въздействието върху мозъка, скелетната система и органите на прекомерната загуба на незаменими аминокиселини, минерали, глюкоза и протеини, екскретирани с урината.
Следователно, лечението на синдрома е насочено към следните задачи:
- Максимално възможна корекция на дефицита на калий, бикарбонати, промени в киселинно-алкалния баланс на кръвта за намаляване на ацидозата.
- Терапия на фосфат-диабет (D-резистентен рахит) с акцент върху недопустимостта на ограничаване на течностите.
- Лечение на основното заболяване, което провокира развитието на придобития синдром при възрастни.
медицински
 За намаляване на загубата на фосфор и калций се използват специални препарати с витамин D, това са l,25 (OH) D3 и l (OH) D3.
За намаляване на загубата на фосфор и калций се използват специални препарати с витамин D, това са l,25 (OH) D3 и l (OH) D3.
Начални дози витамин D3 на ден 10-15 хиляди IU. Увеличаването на дозата се извършва постепенно, като се увеличава на всеки 12-14 дни (под контрола на теста Sulkovich и съдържанието на фосфор в кръвта). При липса на признаци на интоксикация и лека екскреция на калций в урината е позволено да се увеличи дозата, като се увеличи до 100-150 хиляди IU на ден и продължи терапията до нормални нива на фосфор и алкална фосфатаза в кръв. Когато стойностите им се стабилизират, дозата не трябва да се повишава допълнително.
Терапията с витамин D се провежда в няколко курса, за да се предотвратят кризи от прогресия на рахитичните деформации на костите.
Най-добрият вариант е използването на активни метаболити D3 - Oxidevit (0,5 - 1,5 mcg на ден), калциотриол (Rocaltrol).
Те включват калциеви препарати (калциев глюконат на ден до 1,5 - 2 грама), фосфор (0,5 - 1 грам на ден), фитин.
От неорганичните фосфати се използва сместа на Олбрайт, като се приема по 1 голяма лъжица 4 до 5 пъти на ден. Фосфатите се използват под формата на разтвор и таблетки в дози, изчислени на 10 mg на килограм телесно тегло, 4 пъти на ден (винаги с препарати от витамин D, за да се избегне хиперпаратироидизъм).
При изразен калиев дефицит се използват Panangin, Asparkam.
За всяко назначение постоянно се наблюдава CBS или киселинно-алкалното състояние на кръвта. Обикновено кръвта има леко алкална реакция, а рН е в диапазона 7,35 - 7,45. При ацидоза, когато стойността на pH падне под 7,35, кръвта придобива повишена киселинност.
В тези случаи е показана интравенозна инфузия на 4% разтвор на натриев бикарбонат или пиене на разтвор (50-60 ml на ден), който включва лимонена киселина - 2 g, натриев цитрат - 3 g, калиев цитрат - 3,3 g, вода. 100 мл. 1 ml от тази алкализираща смес съдържа 1 mmol натрий и калий. Високата киселинност на кръвта също се неутрализира с помощта на сода бикарбонат (натриев бикарбонат).
Въз основа на практически резултати, някои експерти препоръчват Unithiol като средство за повишаване на активността на тиол-зависимите ензими.
Когато се предписва цистиноза: дитиотрентал в размер на 25 mg на килограм тегло на пациента след 3 часа; Цистеамин в дневна доза от 90 mg/kg.
Има положителни резултати от употребата на пенициламин, който понижава концентрацията на пирогроздена киселина в кръвта, намалява степента на екскреция на аминокиселини и насърчава растежа на алкалните резерви в организма.
Хормоналните анаболи, включително метилтестостерон, имат добър ефект върху функционирането на бъбречните тубули.
Лечението в болница е показано при изразени метаболитни нарушения, включително хипо- и хипергликемия, скелетни деформации.
Предотвратяване
Съвременната профилактика на вроден синдром на Fanconi при наличие на подобна патология в семейството се състои в предварително генетично консултиране. Рискът от развитие на заболяването за така наречените братя и сестри, тоест сестри и братя, е около 25%.
При вторичната форма на синдрома симптомите намаляват или напълно изчезват с активно лечение на основното заболяване или придобитото патологично състояние.
Прогноза
Прогнозата на заболяването зависи от формата (първична, вторична) на синдрома, тежестта на проявите и началото на лечението.
Например, симптомите на придобит синдром изчезват, когато се елиминира провокативната причина.
При вродена патология, на фона на липсата на цистинови отлагания в тъканите, ходът на заболяването не представлява сериозна заплаха за живота, в противен случай, особено без подходящо лечение, смъртта на пациента се прогнозира до 10-20 години от нарастваща бъбречна недостатъчност.
Въпреки това, дори при тежки промени в бъбреците: пиелонефрит, тубулоинтерстициален нефрит, бъбречна недостатъчност, с ранно лекарствено излагане на патологичния процес и продължителна терапия, се установява известен баланс в хомеостазата, при който прогнозата за нормално качество на живот за десетилетия е добре.
В медицинската практика има истории на случаи, когато при деца на 7-8 години наследственият синдром на Fanconi е практически "спрян" с настъпването на дългосрочна ремисия, ясно подобрение на състоянието на детето и дори възстановяване.
В случай на нарушение на бъбречната функция, отговорна за обратната абсорбция на хранителни вещества, се открива синдром на Fanconi. Второто име на състоянието е синдром на de Toni-Debre-Fanconi. При патология се наблюдават глюкоза, протеин c, възникват метаболитни нарушения. Заболяването се предава по наследство. Диагнозата се основава на клинични находки. Целите на лечението са компенсиране на дефицитни състояния, елиминиране на бъбречна недостатъчност.
При деца сайдерът de Toni-Debre-Fanconi причинява рахит, изоставане в физическото развитие, слабост на мускулната тъкан.
Описание и причини
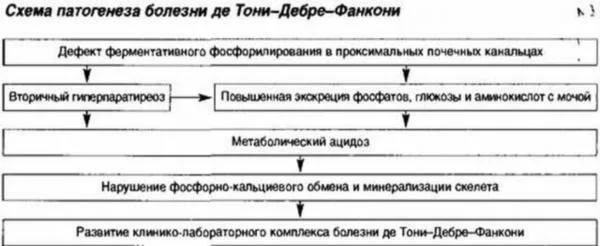
Болестта на De Toni-Debre-Fanconi се проявява с тежка тубулна дисфункция, в резултат на което се нарушава процесът на обратна абсорбция на вещества и йони, полезни за тялото. В същото време се увеличава освобождаването на бикарбонати, откриват се общи метаболитни неуспехи. Особености:
- спад в съдържанието на калций с фосфор в кръвната плазма;
- нарушение на киселинно-алкалния баланс, което се проявява чрез ниска киселинност и бикарбонати в кръвта;
- нормална екскреция на калций под формата на краен метаболит в урината с повишени скорости на естествено почистване на тялото от фосфати;
- патологично висока активност на алкалната фосфатаза;
- появата на захар в урината;
- общо нарушение на производството и екскрецията на продуктите от синтеза на аминокиселини;
- увеличени обеми на отделена урина с намалена плътност.
 Болест на Тони-Дебре-Фанкони при деца.
Болест на Тони-Дебре-Фанкони при деца. Синдромът на Fanconi се развива както при деца, така и при възрастни. Има вродени и придобити видове патология. Според тежестта на симптомите, както и вида и тежестта на метаболитните нарушения се разграничават 2 форми на заболяването:
- Първият тип се проявява под формата на изразено изоставане във физическото развитие с ясна деформация на костите на скелета, тежка клинична картина, чести фрактури на фона на остра липса на калций поради лошо усвояване на веществото в червата.
- Вторият вариант се проявява под формата на умерена степен на изоставане във физическото развитие, лека проява на симптоми със слабо разрушаване на костите и достатъчно ниво на калций.
Причини за заболяването
Провокиращите фактори са разделени на тези, които причиняват придобитата форма на синдрома на de Toni-Debre-Fanconi, и тези, които са отговорни за появата на вродена патология. Следните състояния се признават за провокатори на вторично нарушение:
- непоносимост към фруктоза;
- липса на клетъчни ензими;
- хронично отравяне с токсини;
- тежък дефицит на витамин D.
Самият синдром се причинява от една лоша наследственост или нова мутация на гена, отговорен за метаболитните процеси. Синдромът на De Toni-Debre-Fanconi често се развива не сам, а заедно с цистиноза, галактоземия, синдром на Wilson, първична тирозинемия и непоносимост към фруктоза. По-често тази форма се фиксира при деца, отколкото при възрастни.
Провокиращи фактори
 Острият дефицит на витамин D може да провокира заболяването.
Острият дефицит на витамин D може да провокира заболяването. Синдромът на Fanconi е по-вероятно да се появи при хора със следните наследствени заболявания:
- неизправност в метаболитните процеси на цистин (аминокиселини в състава на протеините);
- непоносимост към млечни продукти (дори кърма);
- дефекти в различни ензими, отговорни за синтеза и разграждането на гликоген;
- неуспех в обмяната на ароматни аминокиселини;
- непоносимост към фруктоза;
- проблеми с метаболизма на медта (болест на Коновалов-Уилсън);
- нарушение на метаболизма на миелина и дисфункция на ензима сулфатаза;
- постоянно излагане на токсини върху тялото (лекарства, отрови, тежки метали);
- неуспех в метаболитните процеси, включващи протеин, което води до отлагането на специфичен комплекс - амилоид;
- тежък дефицит на витамин D.
вродени и придобити
Синдромът на Fanconi е тежко рахитоподобно заболяване, което може да бъде първично (вродено) или вторично (придобито).
Вродената патология на Тони-Дебре-Фанкони е наследствена, т.е. предава се от родители на потомци. Често се развива във връзка с други генетични заболявания. Състоянието се характеризира с остро нарушение на метаболитните процеси, в резултат на което, наред с дисфункцията на бъбречните тубули, се развива слепота, синдром на увеличен черен дроб, синдром с постоянно намаляване на концентрацията и тъканната активност на тиреоидните хормони. Механизмът на наследяване на синдрома се определя от вида на патологията, с която се развива.
Придобитият синдром на de Toni-Debre-Fanconi често се провокира от различни лекарства:
- лекарства за химиотерапия, използвани при лечението на рак;
- антиретровирусни лекарства;
- остарял тетрациклин.
Провокаторите на развитието на вторични форми на патология са сложна бъбречна трансплантация, онкологични заболявания на кръвта, тежки дефицитни състояния (главно с липса на витамин D). Ако вродено заболяване се развива вътреутробно и се фиксира главно при деца, тогава вторичната форма може да се развие и при възрастни.
Механизъм на развитие и протичане
 Реабсорбцията на веществата в бъбреците е нарушена.
Реабсорбцията на веществата в бъбреците е нарушена. Синдромът на De Toni-Debre-Fanconi се причинява от вродена мутация на гена, отговорен за метаболитните процеси, но са отбелязани случаи на нова трансформация, причинена от други патологии. Поради малабсорбция на много полезни вещества и йони се развиват тежки дефицитни състояния:
- при липса на аминокиселини се появява тежка дистрофия, растежът и физическото развитие се забавят;
- при отстраняване на фосфор и бикарбонати - неуспех на процеса на минерализация с началото на разрушаването на костната тъкан;
- с натрупване на захар в урината - нарушение на регулацията на въглехидратния метаболизъм;
- когато калий се екскретира с урината, мускулна атрофия, спад на кръвното налягане до 80 mm Hg. Изкуство. и по-долу;
- с мащабни метаболитни нарушения - разрушаване на бъбречната тъкан (стесняване на лумена с изравняване на епитела и скъсяване на тубулите, в които се извършва реабсорбция).
Симптоми при деца и възрастни
Първите прояви на вроден синдром на Тони-Дебре-Фанкони се появяват през първата година от живота, по-рядко - от 1,5 години. Бебето често уринира, има субфибралност (до 37-38 ° C), запек, повръщане. До 6-годишна възраст, ако не се лекуват, децата не ходят, а до 12-годишна възраст бъбреците отказват, което е изпълнено със смърт. Пациенти - необщителни, известни, срамежливи. Умствените и когитативните функции са нормални. Последиците от синдрома са проблеми с Народното събрание и зрението, развитие на хроничен имунен дефицит, дисфункция на пикочно-половите органи и стомашно-чревния тракт.
Симптомите при деца се развиват в по-голямата си част поради дефицит на фосфат (като фосфатен диабет) и се проявяват чрез:
- нестабилност на походката;
- нисък ръст;
- ниска мобилност;
- закръгленост на краката;
- изкривяване на костите на скелета (по-специално на гръбначния стълб);
- болка в костите, защо бебето не иска да ходи;
- рязко увеличаване на обема на отделената урина;
- спад в специфичното тегло на фона на качествена промяна в състава на урината;
- мускулна атония;
- болка в костите;
- хипертонична болест;
- хронична бъбречна недостатъчност (при липса на лечение);
- остеопороза, остеопения на фона на намаляване на костната плътност.
Болестта на De Toni Debre Fanconi, обикновено наричана синдром на Fanconi, е свързана с нарушена бъбречна функция. Неизправностите, които възникват в човешкото тяло поради това заболяване, водят до усложнения. Това е последвано от статия, чиято информация ще помогне на читателя да разбере характеристиките на заболяването.
Какво представлява болестта на Фанкони
 Името на болестта идва от името на швейцарския лекар Фанкони, който в началото на миналия век пръв изучава причините и симптомите на болестта. Фанкони изследва ниски деца с рахит, които имат промени в урината. Лекарят събра тези прояви заедно и ги постави в една патология - синдром на Фанкони.
Името на болестта идва от името на швейцарския лекар Фанкони, който в началото на миналия век пръв изучава причините и симптомите на болестта. Фанкони изследва ниски деца с рахит, които имат промени в урината. Лекарят събра тези прояви заедно и ги постави в една патология - синдром на Фанкони.
Наблюдавайки пациентите в продължение на две години, Тони добави още 2 патологични фактора към съществуващите патологични фактори: намаляване на нивото на фосфатите в кръвта под установената норма и хипераминоацидурия.
Патологията се предава по наследство. Урината на пациента по време на анализа е пълна с излишно количество захар, протеини и фосфати. Метаболизмът също се забавя и усвояването на бъбречните продукти в кръвта спира.
В света на всеки 350 хиляди новородени има 1 бебе с горния синдром. Струва си да се отбележи, че възрастните рядко се разболяват от този дефект.
Причини за това заболяване
Днес причините за синдрома на Fanconi могат да бъдат различни. Лекарите предполагат, че развитието на болестта е свързано с необяснима генна мутация, по време на която се променят функциите на ензимите, съдържанието на фосфор в кръвта намалява и работата на аминокиселините се нарушава. Някои са склонни да заключат, че патологията се причинява от дисфункция на протеините на тубулите на бъбреците, която се развива поради дефекти в структурата на митохондриите.
Други причини за синдрома:
- отравяне с отрови и токсини, които се появяват в резултат на поглъщане на тежки метали;
- дефицит на витамин D, причиняващ дефекти на генетично ниво;
- неуспехи в асимилацията на необходимите за тялото клетъчни ензими;
- отлагане в тъканите на амилоид по време на нарушение на протеиновия метаболизъм;
- нарушение на метаболитния процес в тялото, наречено цистиноза;
- тирозинемия.
Много изследвания също показват, че заболяването е едно от усложненията на рахита. Рахитът се появява поради липса на клетъчни ензими, докато повечето от полезните и необходими вещества напускат човешкото тяло с урината, превръщайки костната тъкан в омекотена маса.
Точните причини за появата на синдрома на Fanconi, следвайки информацията по-горе, не са идентифицирани, поради което в медицината патологията се нарича глюкофосфаминов диабет.
Симптоми, показващи съществуваща патология
Ярко изразена проява на патология се забелязва в детството. От момента на раждането на бебето и до една година започват да се появяват първите признаци:
- увеличен обем на уринните секрети -;
- запушване;
- появата на запек;
- повишаване на телесната температура;
- постоянна жажда - полидипсия;
- мускулна слабост;
- метеоризъм - газове в червата;
- остра болка в костите, усещайки, че детето непрекъснато плаче, може да бъде объркано с.
Болестта на Де Тони Дебре Фанкони при деца намалява интелектуалните способности. Според физическите данни бебето изостава от връстниците си. Това се дължи на липсата на витамини, които се отделят от тялото на бебето с урината. Долните крайници стават подобни поради извивката на буквата "X". Органите на детето намаляват по време на развитието на атрофия на мускулните влакна. Всичко завършва с факта, че на 5-годишна възраст детето не може да направи малка крачка.
Ненавременното лечение води до факта, че при израстване при юноши функционалността на бъбреците намалява. Има и странични ефекти на заболяването: намалено зрение, сърцето и кръвоносните съдове изпитват повишено натоварване, увреждане на централната нервна система.
Известни форми на идентифицирано заболяване
Болестта е разделена на 2 форми:
- Първичната форма, когато заболяването е наследено от бебето.
- Вторична форма, когато човек се разболя в зряла възраст.
При наследствена патология се откриват дефекти в Х-хромозомата. Мутацията се предава по рецесивен и доминантен тип. За специалистите е трудно да направят точна прогноза. Диагнозата на заболяването е възможна само по време на кърмене. По друг начин първичната форма на синдрома се нарича „инфантилна“.
Степента, до която Х-хромозомата е мутирала, обяснява допълнителни признаци. Можете точно да идентифицирате заболяването по 3 основни дефекта:
- аминоацидурия.
Важно е да се знае! Ако се открият само 2 компонента на горните нарушения, диагнозата се нарича непълна.
Методи за диагностика на заболяването
По принцип синдромът на Fanconi може да бъде открит с помощта на рентгенови лъчи и кръв. Рентгеновата снимка показва следното:
- забавяне на растежа при деца;
- бавно сливане на крайници с фрактури;
- rachiocampsis;
- костна деформация;
- изтощение в тръбните кости;
- в зоните на растеж на костните крайници се образува трошливост;
- намаляване на костната плътност, при което те стават крехки.
С кръвни тестове патологията може да бъде диагностицирана от следните фактори:
- излишък на калий;
- повишени нива на паратироидния хормон;
- ниска концентрация на фосфор и калций;
- повишаване на киселинността в организма;
- ензимът алкална фосфатаза се повишава.
Анализът на урината показва синдром на Fanconi в случай на:
- излишък на фосфатни соли в урината;
- натуриурия;
- повишаване на нивото на аминокиселини в урината;
- повишена глюкоза.
За бележка! В някои случаи за диагностика се използва радиоизотопно изследване, нефробиопсия и биопсия на костна тъкан.
Начини за лечение на този проблем
Лекарите се отнасят сериозно към лечението на патологията. В комплекса се провежда терапевтична терапия: с помощта на лекарства и хирургични интервенции. Често към това се добавя диета. При лечението основната задача на лекаря е да коригира липсата на фосфор в кръвта и да коригира дефицита на калий.
Диета без аминокиселини
Пациентът трябва да спазва диета, за да намали интензивността на отделянето на аминокиселини от тялото заедно с урината. Следователно менюто ще се състои от следните продукти:
- картофи;
- зеле;
- млечни продукти;
- плодове и сокове от тях;
- стафиди;
- фурми;
- сини сливи;
- сушени кайсии.
Лекарят може да предпише на пациента допълнителен прием на витаминни лекарства, богати на витамин D и калий.
Хирургическа интервенция
Пациентът лежи под ножа на хирурга в случай на тежки дефекти в костната тъкан, поради което е невъзможно да се правят прости движения. Операцията е възможна само когато е настъпила фазата на спокойствие и наблюдението на ремисия продължава 1,5 години. За облекчаване на проявите на патология на пациента се препоръчва масаж и вани с борови иглички и морска сол.
Лечение с медикаменти
За да се възстанови нормалното съдържание на калций и фосфор в кръвта, пациентът, в допълнение към диетата, приема лекарства, предписани от лекаря. Също толкова важно е да приемате лекарство, чийто активен елемент е витамин D. Първоначалната доза е 10 000 IU на ден.
С течение на времето дозата ще се увеличи до 100 000 IU на ден. Когато тестовете покажат нормално съдържание на фосфор и калций, приемът на витамин D се отменя. При лечението на вторичната форма на заболяването се използват лекарства, съдържащи фитин и калций.
Синдромът на Fanconi, известен също като болест на Tony-Debre-Fanconi, е бъбречно заболяване, при което е нарушена обратната абсорбция на по-голямата част от веществата, което е придружено от глюкозурия (захар в урината), аминоацидурия (протеин), хиперфосфатурия ( фосфати) и се наблюдават изразени метаболитни нарушения в организма. Това състояние при децата води до изоставане в развитието, рахит и мускулна слабост.

При синдрома на Fanconi реабсорбцията на веществата в бъбреците е нарушена.
Причините за това състояние са различни и все още не са уточнени. По правило синдромът на Fanconi се свързва с непоносимост, недостатъчност на редица клетъчни ензимни системи (например фосфоенолпируват карбоксикиназа), хронично отравяне на тялото с токсини, дефицит на витамин D.
Поради разликата във възгледите, същото състояние може да се нарече още "идиопатичен бъбречен синдром на Fanconi" и "D-резистентен рахит". Понякога се среща терминът "глюкофосфаминов диабет" или "бъбречен нанизъм с D-резистентен рахит".
Среща се сравнително рядко, което отчасти обяснява липсата на информация за този синдром. Средно синдромът се среща при едно на 350 000 бебета.
Разновидности на синдрома на Fanconi

На фона на фосфатния дефицит пищялите придобиват О-образна форма
Разграничават вроден (първичен, идиопатичен) синдром и придобит.
Първичният синдром на Фанкони се счита за Х-свързан.
Може да бъде доминиращ или рецесивен, т.е. наследени по различни начини, прогнозирането на присъствието на потомство е доста трудно.
Генетично обусловеният синдром на Fanconi може да бъде пълен и непълен, т.е. понякога се появяват само 2 от 3 класически симптома (глюкозурия, аминоацидурия, хиперфосфатурия).

Първичният синдром на Fanconi е генетично заболяване
Вторичното, като правило, е следствие от тирозинемия от първия тип. Допринасят за появата на синдрома наследствени патологии на бъбреците. Синдромът на Fanconi доста често се развива след органа (с ниска хистосъвместимост).
Болестта на Тони-Дебре-Фанкони може да бъде резултат от отравяне с живак, уран, олово или кадмий. Понякога се развива при работници в химическото производство, а именно при контакт с толуен, лизол и малеинова киселина.
Понякога синдромът на Fanconi се развива по време на лечение с платинови лекарства, гентамицин и тетрациклинови лекарства с изтекъл срок на годност.
Симптоми на синдрома на Fanconi
Галерия от някои симптоми
 Често уриниране
Често уриниране  Rachiocampsis
Rachiocampsis  Повишена температура
Повишена температура
При децата, като правило, основната част от симптомите са свързани с дефицит на фосфат (подобно на фосфатния диабет). Има трепереща походка ("патица"), нисък ръст, бездействие. Постепенно се формира О-образната форма на пищялите, други кости на скелета (особено гръбначния стълб) се огъват. Поради болката в костите детето ходи малко. Рискът от фрактури поради костна тъкан е много висок.
Децата растат необщителни и срамежливи – не страдат когнитивните (умствени) функции, но се образуват комплекси.

Запекът може също да бъде свързан с болестта на Тони-Дебре-Фанкони.
Първите признаци на заболяването при вродена форма се появяват още през първата година от живота (понякога от 1,5 години).
Детето започва да уринира често, появява се (37-38 0C), развива се запек, може да има повръщане.
С течение на времето родителите забелязват горните симптоми на дефицит на фосфор. До 6-годишна възраст децата обикновено не могат да ходят сами. С развитието на процеса до 12-годишна възраст се формира, което е изпълнено с фатален изход.
Метаболитните нарушения водят до патологии на нервната система, проблеми със зрението, дефекти в развитието на органите на пикочно-половата система, чревни заболявания и хроничен имунен дефицит.
При възрастни се проявява вторичен синдром (често уриниране), обща слабост, мускулна хипотония и болки в костите. Активно се формира бъбречна недостатъчност, развива се артериална хипертония.
Най-лошото от всичко е, че придобитият синдром на Fanconi засяга жени в постменопауза. В допълнение към естественото намаляване на костната плътност (остеопения, остеопороза) се добавя костна чупливост поради дефицит на минерали. Тази ситуация завършва с компресионни фрактури на телата на прешлените, фрактури на главата на бедрената кост и увреждане на пациента.

Остеопорозата е спомагателен симптом при диагностицирането на синдрома на Fanconi
Диагностика на синдрома на Fanconi
Синдромът е рядък, опитен лекар може да го подозира поради рентгеново изследване на костите и някои напреднали биохимични параметри на кръвта и урината.

Появата на шпори е свързана с нарушение на процеса на образуване на кост
Синдромът на Fanconi често се комбинира със системна остеопороза (в този случай остеопорозата е спомагателен симптом). Костната тъкан има груба влакнеста структура, много често се виждат костни израстъци („шпори“). Денситометрията се използва за определяне на костната плътност.
Костите са слабо минерализирани, което се установява чрез анализ на биопсия (тъканна проба).
От страна на бъбреците - по време на изследването се определя дистрофия на бъбречните тубули (по тип "лебедова шия"), епителът се разрушава и бъбречните структури се заменят със съединителна тъкан.
Единственото, което спасява пациентите, е, че гломерулният слой е последният, който се включва в процеса и до самия край на заболяването функцията на бъбреците някак си, но се изпълнява.
Микроскопията на гломерулния епител показва голям брой митохондрии в клетките.
Лечение на заболяването
Първичният синдром на Fanconi не може да бъде излекуван, тъй като говорим за структурни промени в бъбреците, както и за постоянни, генетично обусловени метаболитни нарушения. Такива пациенти постоянно поддържат нивото на калий, лекуват бъбречна тубулна ацидоза и други дефекти във водно-солевия метаболизъм. Фосфат-диабетът в този случай подлежи на стандартна терапия за това заболяване.
Пациентите трябва да пият много течности през целия ден.
Галерия с някои лечения за синдром на Fanconi
 Капкомери
Капкомери  витамини
витамини  Иглолистни бани
Иглолистни бани  Масаж
Масаж
Вторичният синдром на Fanconi в някои случаи е лечим, особено при успешното елиминиране на заболяването или състоянието, което провокира тази бъбречна патология.
Лекарствена терапия
За компенсиране на нарушенията на метаболизма на фосфора и калция се предписват метаболити на витамини от група D - 1 (OH) D3 или 1,25 (OH) D3. Дозите на витамина се титрират от 10 000 IU до 100 000 IU на ден. Дозировката се избира при постоянен лабораторен контрол на нивото на фосфор и калций в кръвта. Предписват се калциеви препарати и фитин per os.
Такава терапия се провежда периодично, на курсове.
Диетична терапия за синдром на Fanconi
Основният принцип на диетата е ограничаване на отделянето на редица вещества от организма, вкл. сяросъдържащи аминокиселини и фосфор. Диетата включва ограничаване на солта и широко използване на алкализиращи храни.
Необходимо е да се въведе в диетата голямо количество плодови сокове и мляко (с нормална поносимост)
 плодови сокове
плодови сокове  ограничаване на солта
ограничаване на солта  Мляко
Мляко  Сушени плодове
Сушени плодове
Ако нарушенията на опорно-двигателния апарат станат критични, тогава се препоръчва хирургична корекция.
Навременната диагностика на метаболитни нарушения според типа синдром на Fanconi при деца позволява да се избегне бързото увреждане на детето, при възрастни - да се намали рискът от фрактури и увреждане на корените на нервните влакна, да се повиши качеството и живота. очакване. При първите признаци на заболяването (полиурия, промяна в цвета на урината, болки в костите и ставите) се консултирайте с лекар.
Синдромът на Fanconi е системно метаболитно разстройство, при което се открива бъбречна дисфункция. Те губят способността да реабсорбират и връщат в кръвния поток аминокиселините, глюкозата, калциевите и фосфорните соли, екскретирани с урината. Това е вроден или придобит органичен дефект. Това е изключително рядко, 1 случай се среща на 350 хиляди новородени. Тази дисфункция обаче е изключително трудно да повлияе на състоянието на тялото.
Нарича се още синдром на de Toni-Debre-Fanconi, фосфатен диабет, наследствен синдром на Fanconi. Някои учени смятат, че тази патология възниква, когато запасите от АТФ (аденозинтрифосфорна киселина) рязко намаляват в клетките. Други излагат версията, че деформацията на рахитична кост възниква поради дефицит на фосфор, неизправност в киселинно-алкалния баланс или поради и двата фактора. Други пък смятат, че бъбречната дисфункция не се дължи на биохимичен, а на структурен дефект. Все още не е установена точната причина.
С тази патология са засегнати бъбречните проксимални тубули. Концентрацията на калциеви и калиеви съединения в кръвния поток остава в нормалните граници. Реакцията на урината е алкална или неутрална. Поради значителни загуби на въглеводороди се развива ацидоза. Това е опасно състояние, при което има нарушение на киселинно-алкалния баланс и "подкиселяване" на организма. Ацидозата обикновено се причинява от всяко дифузно бъбречно заболяване.
Вродената патология може да има различна степен на тежест. При наличие на 3 биохимични дефекта се диагностицира пълна, а при 2 - непълна бъбречна дисфункция. Някои учени смятат, че наследственият синдром на Fanconi е независимо заболяване, подобно на рахит. Най-често обаче придружава други вродени патологии. Не може да бъде:
- цистиноза (заболяване, свързано с кристализацията на аминокиселината цистин в тъканите);
- Болест на Уилсън-Коновалов (патология, причинена от отлагания на мед в бъбреците, черния дроб, мозъка);
- галактоземия (нарушено превръщане на простата захар в глюкоза);
- Синдром на Lowe (патология, проявяваща се със забавяне на растежа, умственото развитие, катаракта и глаукома);
- тирозинемия (пасивност на чернодробния ензим, който разгражда аминокиселината тирозин);
- непоносимост към фруктоза.
Придобитата дисфункция, кръстена на Фанкони, може да възникне поради такива фактори:
- лекарства, които са токсични за бъбреците (аспирин, гентамицин, тетрациклин с изтекъл срок на годност, антивирусни лекарства цидофовир, диданозин, противотуморни лекарства стрептозоцин, ифосфамид и др.);
- отравяне със соли на тежки метали, други агресивни химически съединения;
- остър дефицит на витамин D;
- мултиплен миелом (рак на кръвта);
- амилоидоза (нарушения на протеиновия метаболизъм);
- трансплантация на бъбрек.
Симптоми на заболяването при деца
Най-изразеният вроден синдром на Fanconi при деца. Неговите симптоми се усещат още през първата година от живота на бебетата. Това:
- полиурия (често уриниране с голямо количество течност, отделена от тялото);
- полидипсия (патологично силна жажда);
- повишена температура;
- конвулсии;
- често повръщане без видима причина;
- продължителен запек;
- кожен обрив;
- подуване на ставите;
- уголемяване на бъбреците, лимфните възли, далака;
- предразположение към инфекции.

Поради ежедневното отделяне на витамини, микроелементи, глюкоза от тялото с течност, детето изостава в умственото и физическото развитие. Костите на краката му се огъват, стават отпуснати, а след това мускулите напълно атрофират. Често децата губят способността си да ходят самостоятелно. До 12-13-годишна възраст се развива хронична бъбречна недостатъчност. Понякога зрението се влошава. В допълнение, при синдрома на Fanconi, симптомите на патологията могат да показват комбинирани вродени дефекти на съдовете и сърцето, храносмилателните органи и пикочно-половата система.
Симптомите на заболяването при възрастни
Най-често се отбелязва:
- полиурия;
- хипостенурия (намалена относителна плътност на урината);
- болки в костите;
- остеомалация (омекване на костната тъкан и загуба на сила);
- мускулна слабост;
- ускорено прогресиране на хипертония;
- хронична бъбречна недостатъчност (ако няма лечение).
Полиурията при синдрома на Fanconi при възрастни не е изразена, това е значително различно от излишното количество урина при безвкусен диабет. Най-често този синдром се превръща в проява на множествена миелома или болест на Waldenström (злокачествени лезии на хемопоетичната система). В допълнение към умерената полиурия, нарушената бъбречна активност се проявява чрез намаляване на тяхната концентрационна функция, появата на протеин в урината.
Дали има синдром на Fanconi или не, може да се съди по такива характерни симптоми като висока концентрация на аминокиселини, глюкоза и фосфати в урината. Повишеното отделяне на глюкоза с урината при тази патология се дължи на бъбречна дисфункция. Въпреки това, концентрацията на захар на празен стомах и нивото на хипергликемия при тези пациенти, като правило, са в нормалните граници.
Полиурията винаги е придружена от мускулна слабост, която се усеща най-остро в крайниците. Възниква поради недостиг на калий. В допълнение, почти винаги пациентите са измъчвани от болки в костите.

Симптомите на ацидоза при възрастни и деца са еднакви. Това:
- загуба на апетит;
- безпричинно повръщане;
- запек или диария;
- кисела миризма, идваща от кожата или от устата;
- намаляване на налягането;
- главоболие;
- диспнея;
- безсъние;
- прострация.
Въпреки това лекарите разграничават ацидозата при възрастни от детската. Смята се, че при кърмачета това винаги е вродена патология. При възрастни това може да бъде както по-нататъшно развитие на детска болест, така и придобито усложнение поради увреждане на бъбреците. Симптомите на ацидоза често се комбинират с признаци на интерстициален нефрит и уролитиаза. Бъбречните камъни се образуват поради големи загуби на калций в урината. В тежки случаи се откриват симптоми на остеопороза и остеомалация.
Диагностика на патология
Идентифицирането на синдрома на de Toni-Debre-Fanconi започва със събиране на анамнеза, преглед на пациента и лабораторни изследвания. Характерни признаци на патология в биохимичната кръвна картина са ниските нива на калций, фосфор, калий и натрий, както и глюкоза, аланин, глицин, глутаминова киселина. Но в урината - високо ниво на фосфор и глюкоза, има белтък и левкоцити.
При значителна загуба на бикарбонати в урината и прекомерно натрупване на киселини в организма се диагностицира метаболитна ацидоза. Почти всички пациенти имат излишък на нивото на пирогроздена и млечна киселина в кръвта. Открива се и спад в активността на ензимите на енергийния метаболизъм.
При инструментална диагностика е задължително ултразвуково изследване на бъбреците и уретерите. Нефробиопсията (изследване на миниатюрни проби от бъбреците) ви позволява да видите деформацията на проксималните тубули, които са удължени като лебедова шия. В по-късните стадии на заболяването се открива атрофия на бъбречните гломерули.

Рентгенографията на деформираните долни крайници помага да се открие дегенерацията на костната тъкан. При деца често се откриват скрити фрактури на епифизния хрущял, поради което костите впоследствие спират да растат по дължина и стават асиметрични. В тибията се откриват неоплазми, наподобяващи шпори.
В по-късните етапи от развитието на синдрома се диагностицира остеопороза. Рискът от фрактури на тръбните кости е висок. Стадият на остеопорозата се определя чрез рентгенова денситометрия. Ниската минерализация на костните тъкани се открива при изследването на техните проби, също получени чрез биопсия.
Основните диагностични критерии:
- Значителен дефицит в теглото и височината на детето.
- Слабост на статично-моторните функции.
- Рахитоподобни деформации на скелета (разрушаването на структурата на костната тъкан се потвърждава от рентгенови лъчи).
- електролитни нарушения.
При синдром на Fanconi при възрастни лекарите трябва да проведат диференциална диагноза, за да изключат патологии, подобни на него. Това:
- наследствени заболявания (цистиноза, галактоземия, болест на Уилсън-Коновалов, тирозинемия и др.);
- хроничен пиелонефрит;
- диабет;
- вторичен хиперпаратироидизъм;
- множествена миелома;
- отравяне с лекарства, агресивни химикали;
- обширни изгаряния.
Лечение на патология
В идеалния случай, когато синдромът на Fanconi се лекува от генетик, хематолог, но не всяко медицинско заведение разполага с такива специалисти. Най-често тази патология се занимава с нефролог или уролог. При установяване на признаци на хиперпаратироидизъм (хиперсекреция на паращитовидните жлези) е необходима консултация с ендокринолог. При влошаване на зрението е необходимо да се прегледа офталмолог.

Тактиката на лечение включва:
- Попълване на електролитен дефицит.
- Премахване на нарушения на киселинно-алкалния баланс.
- Терапия за бъбречна недостатъчност.
- Премахване на болезнени симптоми.
За медицинско лечение се използват:
- Калцитриол, Оксидевит и други препарати с витамин D;
- калциев глюконат;
- Фитин, калциев глицерофосфат, алуминиев фосфат;
- алкализиращи разтвори Bizitra, Polizitra;
- Индометацин, Метилтестостерон, Хипотиазид (с тежко увреждане на проксималните тубули);
- Панангин, Аспаркам;
- Цистин, Меркаптамин;
- антибиотици;
- кортикостероиди и др.
Тъй като синдромът на Fanconi е хроничен, лечението се провежда дълго време, в големи курсове след задължителни прекъсвания. Често е възможно метаболизмът да се доближи до нормата, да се облекчи тежестта на проявата на заболяването и да се предотвратят опасни усложнения. Но е изключително трудно да се отървете напълно от синдрома на de Toni-Debre-Fanconi, той често дава рецидиви.
Терапията на синдрома е насочена към елиминиране на ацидозата, попълване на запасите от калий, калциев бикарбонат, фосфати и други електролити. Най-важна роля в това играе терапевтичната диета. Трябва да пиете много вода и да намалите количеството сол. Яжте малки порции, но често. Въглехидратите трябва да бъдат строго дозирани, за да няма излишна глюкоза в кръвта.
Подобни статии
-
Когато съпругът е против дете, как да забременеете без негово знание?
Понякога можете да забременеете по небрежност. За да не се случи това, е важно да знаете как можете да заченете дете случайно и какви средства можете да използвате, за да избегнете нежелана бременност. Също така в тази статия можете да намерите информация за...
-
Какви камъни и амулети са подходящи за Телец според хороскопа и датата на раждане Талисман на слон за Телец
Април-май Телците (21 април - 20 май) са премерени, не са суетливи и колосално продуктивни! Тяхната завидна упоритост може да доведе другите до дръжката, но те знаят точно какво правят и защо имат нужда от това. Сред положителните...
-
Ограничения за достъп до данни в роли 1c
Всички настройки на потребителските права, които ще направим в рамките на тази статия, се намират в раздел 1C 8.3 „Администриране“ - „Настройки на потребител и права“. Този алгоритъм е подобен в повечето конфигурации на ...
-
1c стартира тънък клиент вместо дебел
Платформи: 1C: Enterprise 8.3, 1C: Enterprise 8.2, 1C: Enterprise 8.1 Конфигурации: Всички конфигурации2012-11-16 21362 Те се стартират чрез указване на специални ...
-
Доказателства за известни начини за кражба на ток Как да разберете кой краде ток
Повишаването на енергийните тарифи е една от поразителните характеристики на задълбочаващата се икономическа криза. В този контекст кражбата на електроенергия и проблемите, свързани с разкриването й, са от първостепенно значение.Начини за разкриване на кражба ...
-
Характеристики на монтаж на контакти и превключватели на различни повърхности
Поздрави на всички читатели на нашия блог Днес, скъпи читатели, искам да подчертая темата как да инсталирам гнезда. Тази процедура е много често търсена при подмяна на стар контакт с нов в случай на повреда, когато ...