Особенности кинетики ферментативной реакции. Условия проведения ферментативной реакции
Фермент Е обратимо соединяется с субстратом S, образуя нестойкий промежуточный фермент-субстратный комплекс ES, который в конце реакции распадается с освобождением фермента и продуктов реакции Р.
Эти представления легли в основу теории «ключ-замок» Э. Фишера (1890). Структура активного центра комплементарна молекулярной структуре субстрата, обеспечивая тем самым высокую специфичность фермента. В образовании фермент-субстратных комплексов участвуют водородные связи, электростатические и гидрофобные взаимодействия, а в ряде случаев также ковалентные, координационные связи.
Д. Кошлендом была разработана теория «индуцированного соответствия» (1958) . Пространственное соответствие структуры субстрата и активного центра фермента создается в момент их взаимодействия друг с другом, что может быть выражено формулой «перчатка - рука». Субстрат индуцирует конформационные изменения молекулы фермента таким образом, что активный центр принимает необходимую для связывания субстрата пространственную ориентацию. Т.е. фермент только в момент присоединения субстрата будет находиться в активной (напряженной) Т-форме (tensile) в отличие от неактивной R-формы (relaxe).
В настоящее время гипотеза Кошланда постепенно вытесняется гипотезой топохимического соответствия. Сохраняя основные положения теории «индуцированного соответствия», она объясняет специфичность действия ферментов узнаванием той части субстрата, которая не изменяется при катализе.
Подобно другим катализаторам, ферменты, с термодинамической точки зрения, ускоряют химические реакции за счет снижения энергии активации.
Энергией активации называется энергия, необходимая для перевода всех молекул моля вещества в активированное состояние при данной температуре.
Как катализируемая ферментом, так и не катализируемая им реакция имеет одинаковую величину стандартного изменения свободной энергии (ΔG). Однако ферментативная реакция имеет более низкую энергию активации. Действуя на скорость реакции, ферменты не изменяют положения равновесия между прямой и обратной реакциями, а лишь ускоряют его наступление.
2.3. Кинетика ферментативных реакций
Ферментативная кинетика исследует влияние химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температура, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Скорость ферментативной реакции (V) измеряют по убыли количества субстрата или приросту продукта за единицу времени.
При ферментном катализе фермент (Е) обратимо соединяется с субстратом (S), образуя нестойкий фермент-субстратный комплекс (ES), который в конце реакции распадается с освобождением фермента (Е) и продуктов реакции (Р):
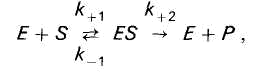
Важная особенность ферментативных реакций – насыщение фермента субстратом . При низкой концентрации субстрата скорость реакции прямо пропорциональна его концентрации. При высокой - скорость реакции максимальна, становится постоянной и не зависящей от концентрации субстрата [S] и целиком определяется концентрацией фермента (рис. 11).
|
|
|
Рис. 11. Зависимость скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента. |

K S – константа диссоциации фермент-субстратного комплекса ES, обратна константе равновесия:
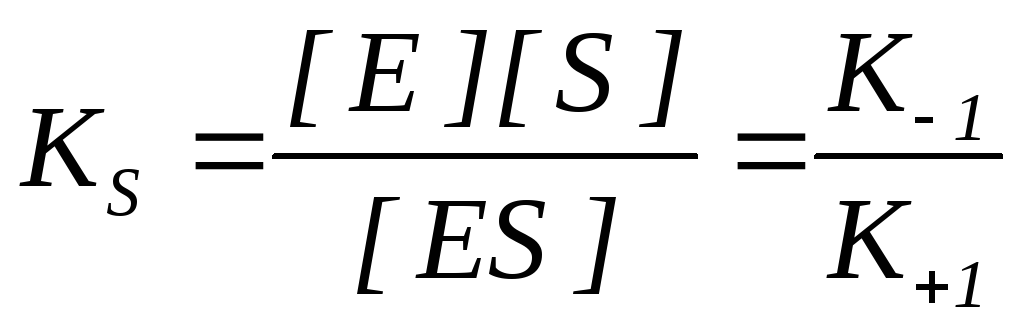
Чем меньше значение K S , тем выше сродство фермента к субстрату.
Количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции выражает уравнение Михаэлиса-Ментен :
 ,
,
- скорость реакции, V max - максимальная скорость ферментативной реакции.
Бриггс и Холдейн усовершенствовали уравнение, введя в него константу Михаэлиса K M , определяемую экспериментально.
Уравнение
Бриггса – Холдейна: 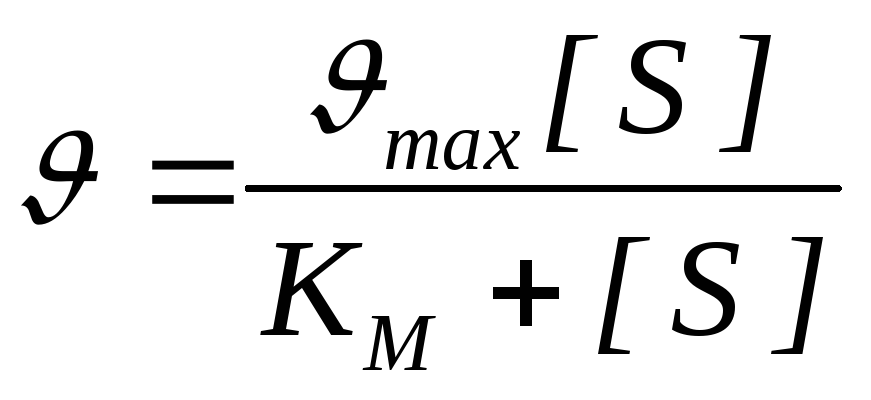
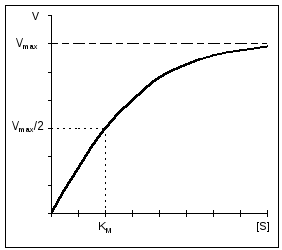
Константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость ферментативной реакции составляет половину от максимальной (рис. 12). К m показывает сродство фермента к субстрату; чем меньше ее значение, тем больше сродство.
Экспериментальные значения К m для большинства ферментативных реакций с участием одного субстрата обычно 10 -2 -10 -5 М. Если реакция обратима, то взаимодействие фермента с субстратом прямой реакции характеризуется К m , отличающейся от таковой для субстрата обратной реакции.
ОБЩАЯ ХАРАКТЕРИСТИКА ФЕРМЕНТОВ
Ферменты – биологические катализаторы.
Химическая природа ферментов. Активный центр ферментов.
Механизм ферментативного катализа.
I . Ферменты – биологические катализаторы белковой природы, способные во много раз ускорять химические реакции, протекающие в организме, но сами не входящие в состав конечных продуктов реакции.
Вещества, на которое действует фермент, называют субстратами.
Все многообразие биохимических реакций, протекающие в микроорганизмах, растениях и животных катализируется соответствующими ферментами. Велика роль ферментов в технологии пищевых продуктов. В основе производства любого пищевого продукта лежат либо биохимические (ферментативные), либо физико-химические процессы, либо эти процессы взаимосвязаны.
В отличие от неорганических катализаторов ферменты имеют свои особенности:
Скорость ферментативного катализа на несколько порядков выше (от 10 3 до 10 9), чем не биологического катализатора;
действие каждого фермента высокоспецифично, т.е. каждый фермент действует только на свой субстрат или группу родственных субстратов;
ферменты катализируют химические реакции в мягких условиях, т.е. при обычном давлении, высокой температуре (20-50С) и при значениях рН среды, в большинстве случаев близких к нейтральной.
С точки зрения локализации ферментов в клетке их подразделяют на внеклеточные и внутриклеточные.
Внеклеточные ферменты выделяются живой клеткой во внешнюю среду,внутриклеточные – находятся либо в клеточных органеллах, либо в комплексе с надмолекулярными структурами.
Особую группу ферментов составляют полиферментные комплексы, в состав которых входит ряд ферментов, катализирующих последовательные реакции превращения какого-либо субстрата. Эти комплексы локализованы во внутримолекулярных структурах таким образом, что каждый фермент располагается в непосредственной близости от фермента, катализирующего реакцию в цепи данной последовательности реакций. Благодаря такому расположению ферментов процесс диффузии субстрата и продуктов реакции сводится к минимуму.
II . Ферменты – высокомолекулярные белковые соединения.
Как и другие белки, ферменты имеют 4 уровня структуры, им присущи все физико-химические свойства белков, и лишь одна отличительная особенность – способность ускорять химические реакции. Ферменты могут быть простыми – однокомпонентными и сложными двухкомпонентными.
Однокомпонентные ферменты – построены из полипептидных цепей и при гидролизе распадаются только до аминокислот.
Двухкомпонентные ферменты – состоят из белковой части – апоформента и небелковой части – кофактора . Оба компонента в отдельности лишены ферментативной активности. Только соединившись вместе (холофермент ) они приобретают свойства, характерные для биокатализаторов. Роль кофактора может выполнять какой-либо ион (Zn 2+ , Mg 2+ , Fe 2+ , Cu 2+ , реже K + и Na +) или органическое соединение (витамины, нуклеотиды). Кофакторы органической природы называются коферментами.
Тип связи между кофактором и апоферментом может быть различным. В некоторых случаях они существуют отдельно и связываются только во время протекания реакции; в других случаях кофактор и апофермент связаны постоянно, иногда прочными, ковалентными связями.
Активный центр ферментов –это локальный участок молекулы фермента, который участвует в акте катализа. Воднокомпонентных ферментахактивный центр образуется в результате определенной ориентации аминокислотных остатков полипептидной цепи. Обычно в его формировании принимает участие небольшое количество аминокислот, в пределах 12-16. Функциональные группы этих аминокислот могут принадлежать звеньям полипептидной цепи, удаленным друг от друга. Их сближение связано с формированием третичной структуры ферменты.
В двухкомпонентных ферментах активный центр представляет собой комплекс кофактора и некоторых примыкающих к нему аминокислотных остатков.
В активном центре различают контактный (якорный ) участок, функция которого – связывать субстрат, икаталитический – где происходит превращение субстрата в продукты реакции после его связывания контактным участком. В формировании этих участков принимают участие следующие функциональные группы: СООН-группы дикарбоновых аминокислот или концевые группы полипептидной цепи; имидазольная группа гистидина; ОН-группа серина;NH 2 - группа лизин и концевые группы полипептидной цепи; фенольная группа тирозина и гидрофобные остатки алифатических аминокислот.
III . Скорость любой ферментативной реакции определяетсяэнергетическим барьером , который необходимо преодолеть реагирующим молекулам. По Аррениусу, химическая реакция с точки зрения энергетики процесса описывается уравнением
N = N 0 e -(E акт /RT) ,
где N– число активных молекул;N 0 - общее число реагирующих молекул; е – основание натурального логарифма;R– газовая постоянная;T– абсолютная температура; Е акт – энергия активации.
Энергия активации – дополнительное количество энергии, необходимое для того, чтобы все молекулы преодолели энергетический барьер реакции и вступили в неё. Эта энергия представляет собой разность общей энергии реагирующих молекул и энергиивозбужденного переходного состояния. Чем больше энергия активации в реагирующей системе, тем выше энергетический барьер и тем ниже скорость реакции.
Важнейшая функция фермента – снижение энергии активации катализируемого процесса. На рис. 1 представлен график изменения энергии неферментативной (1) и ферментативной (2) реакций. Фермент снижает высоту энергетического барьера (Е акт Е акт).
Механизм ферментативного катализа во многом остается пока еще не выясненным. Однако большую роль в создании ферментативной кинетики сыграли работы М. Михаэлиса и М. Ментен, в которых было развито представление о фермент-субстратном комплексе . Образование этого комплекса и ведет к снижению энергии активации реакции.
Процесс ферментативного катализа можно условно подразделить на три стадии:
Стерическое связывание субстрата Sс активным центром фермента Е (образование фермент-субстратного комплекса ЕS).
Преобразование первичного комплекса ЕSв активированный переходный комплекс ЕS ≠ .
Отделение конечного продукта Р реакции от фермента.
Первая стадия непродолжительна по времени и зависит от концентрации субстрата и фермента в среде, от скорости диффузии субстрата к активному центру фермента. В образовании комплекса ЕSмогут участвовать в различных сочетаниях как ковалентные, координационные, ионные связи, так и менее прочные формы связей - электростатическое притяжение полярных групп, ван-дер-ваальсовы силы сцепления между неполярными участками молекул, водородные связи. Характер этих связей обусловлен химическими особенностями и субстрата, и функциональных групп, входящих в активный центр фермента.
Вторая стадия является, собственно, актом катализа, т.е. актом разрыва или образования в субстрате новых связей; она наиболее медленная и лимитирует скорость протекания химической реакции. На этой стадии и происходит снижение энергии активации ферментативной реакции, за счет образования активного переходного комплекса ЕS ≠ .
На молекулярном уровне более четкое представление о механизме действия ферментов дает теория кислотно-основного катализа. Любая реакция, идущая с разрывом ковалентных связей, предполагает участие двух противоположных по характеру электронных компонентов. Электроны разрываемой связи должны оттягиваться к электро-фильному компоненту и уходить от нуклеофильного. Реагенты, которые могли бы обусловить такую электронную перестройку - это кислота и основание. Однако в одном и том же растворе создать одновременно высокие концентрации обоих компонентов невозможно, поскольку они нейтрализуют друг друга. В белковой молекуле фермента благодарязакреплению на каталитической площадке электрофильных и нуклеофильных групп не происходит прямой реакции нейтрализации. Это, собственно, и определяет акт катализа. Находясь на определенном расстоянии друг от друга, электрофильные и нуклеофильные группы каталитического участка фермента не только связываются с реагирующими группами субстрата, но и оказывают сильное поляризующее действие на группы субстрата. К этому следует добавить возможность флуктуации зарядов в комплексе ЕS, которая создает высокую степень эффективности данной поляризации. Это и является причиной снижения величины энергии активации при ферментативном катализе.
В соответствии с теорией ковалентного катализа некоторые ферменты взаимодействуют со своими субстратами, образуя нестабильные, ковалентно связанные фермент-субстратные комплексы. Из этих комплексов в ходе последующей реакции образуются продукты реакции, причем значительно быстрее, чем в случае некатализируемых реакций.
Таким образом, третья стадия, завершающаяся образованием продуктов реакции, обеспечивается процессами, протекающими на предыдущих стадиях.
ОБЩАЯ ХАРАКТЕРИСТИКА ФЕРМЕНТОВ
Ферменты – биологические катализаторы.
Химическая природа ферментов. Активный центр ферментов.
Механизм ферментативного катализа.
I . Ферменты – биологические катализаторы белковой природы, способные во много раз ускорять химические реакции, протекающие в организме, но сами не входящие в состав конечных продуктов реакции.
Вещества, на которое действует фермент, называют субстратами.
Все многообразие биохимических реакций, протекающие в микроорганизмах, растениях и животных катализируется соответствующими ферментами. Велика роль ферментов в технологии пищевых продуктов. В основе производства любого пищевого продукта лежат либо биохимические (ферментативные), либо физико-химические процессы, либо эти процессы взаимосвязаны.
В отличие от неорганических катализаторов ферменты имеют свои особенности:
Скорость ферментативного катализа на несколько порядков выше (от 10 3 до 10 9), чем не биологического катализатора;
действие каждого фермента высокоспецифично, т.е. каждый фермент действует только на свой субстрат или группу родственных субстратов;
ферменты катализируют химические реакции в мягких условиях, т.е. при обычном давлении, высокой температуре (20-50С) и при значениях рН среды, в большинстве случаев близких к нейтральной.
С точки зрения локализации ферментов в клетке их подразделяют на внеклеточные и внутриклеточные.
Внеклеточные ферменты выделяются живой клеткой во внешнюю среду,внутриклеточные – находятся либо в клеточных органеллах, либо в комплексе с надмолекулярными структурами.
Особую группу ферментов составляют полиферментные комплексы, в состав которых входит ряд ферментов, катализирующих последовательные реакции превращения какого-либо субстрата. Эти комплексы локализованы во внутримолекулярных структурах таким образом, что каждый фермент располагается в непосредственной близости от фермента, катализирующего реакцию в цепи данной последовательности реакций. Благодаря такому расположению ферментов процесс диффузии субстрата и продуктов реакции сводится к минимуму.
II . Ферменты – высокомолекулярные белковые соединения.
Как и другие белки, ферменты имеют 4 уровня структуры, им присущи все физико-химические свойства белков, и лишь одна отличительная особенность – способность ускорять химические реакции. Ферменты могут быть простыми – однокомпонентными и сложными двухкомпонентными.
Однокомпонентные ферменты – построены из полипептидных цепей и при гидролизе распадаются только до аминокислот.
Двухкомпонентные ферменты – состоят из белковой части – апоформента и небелковой части – кофактора . Оба компонента в отдельности лишены ферментативной активности. Только соединившись вместе (холофермент ) они приобретают свойства, характерные для биокатализаторов. Роль кофактора может выполнять какой-либо ион (Zn 2+ , Mg 2+ , Fe 2+ , Cu 2+ , реже K + и Na +) или органическое соединение (витамины, нуклеотиды). Кофакторы органической природы называются коферментами.
Тип связи между кофактором и апоферментом может быть различным. В некоторых случаях они существуют отдельно и связываются только во время протекания реакции; в других случаях кофактор и апофермент связаны постоянно, иногда прочными, ковалентными связями.
Активный центр ферментов –это локальный участок молекулы фермента, который участвует в акте катализа. Воднокомпонентных ферментахактивный центр образуется в результате определенной ориентации аминокислотных остатков полипептидной цепи. Обычно в его формировании принимает участие небольшое количество аминокислот, в пределах 12-16. Функциональные группы этих аминокислот могут принадлежать звеньям полипептидной цепи, удаленным друг от друга. Их сближение связано с формированием третичной структуры ферменты.
В двухкомпонентных ферментах активный центр представляет собой комплекс кофактора и некоторых примыкающих к нему аминокислотных остатков.
В активном центре различают контактный (якорный ) участок, функция которого – связывать субстрат, икаталитический – где происходит превращение субстрата в продукты реакции после его связывания контактным участком. В формировании этих участков принимают участие следующие функциональные группы: СООН-группы дикарбоновых аминокислот или концевые группы полипептидной цепи; имидазольная группа гистидина; ОН-группа серина;NH 2 - группа лизин и концевые группы полипептидной цепи; фенольная группа тирозина и гидрофобные остатки алифатических аминокислот.
III . Скорость любой ферментативной реакции определяетсяэнергетическим барьером , который необходимо преодолеть реагирующим молекулам. По Аррениусу, химическая реакция с точки зрения энергетики процесса описывается уравнением
N = N 0 e -(E акт /RT) ,
где N– число активных молекул;N 0 - общее число реагирующих молекул; е – основание натурального логарифма;R– газовая постоянная;T– абсолютная температура; Е акт – энергия активации.
Энергия активации – дополнительное количество энергии, необходимое для того, чтобы все молекулы преодолели энергетический барьер реакции и вступили в неё. Эта энергия представляет собой разность общей энергии реагирующих молекул и энергиивозбужденного переходного состояния. Чем больше энергия активации в реагирующей системе, тем выше энергетический барьер и тем ниже скорость реакции.
Важнейшая функция фермента – снижение энергии активации катализируемого процесса. На рис. 1 представлен график изменения энергии неферментативной (1) и ферментативной (2) реакций. Фермент снижает высоту энергетического барьера (Е акт Е акт).
Механизм ферментативного катализа во многом остается пока еще не выясненным. Однако большую роль в создании ферментативной кинетики сыграли работы М. Михаэлиса и М. Ментен, в которых было развито представление о фермент-субстратном комплексе . Образование этого комплекса и ведет к снижению энергии активации реакции.
Процесс ферментативного катализа можно условно подразделить на три стадии:
Стерическое связывание субстрата Sс активным центром фермента Е (образование фермент-субстратного комплекса ЕS).
Преобразование первичного комплекса ЕSв активированный переходный комплекс ЕS ≠ .
Отделение конечного продукта Р реакции от фермента.
Первая стадия непродолжительна по времени и зависит от концентрации субстрата и фермента в среде, от скорости диффузии субстрата к активному центру фермента. В образовании комплекса ЕSмогут участвовать в различных сочетаниях как ковалентные, координационные, ионные связи, так и менее прочные формы связей - электростатическое притяжение полярных групп, ван-дер-ваальсовы силы сцепления между неполярными участками молекул, водородные связи. Характер этих связей обусловлен химическими особенностями и субстрата, и функциональных групп, входящих в активный центр фермента.
Вторая стадия является, собственно, актом катализа, т.е. актом разрыва или образования в субстрате новых связей; она наиболее медленная и лимитирует скорость протекания химической реакции. На этой стадии и происходит снижение энергии активации ферментативной реакции, за счет образования активного переходного комплекса ЕS ≠ .
На молекулярном уровне более четкое представление о механизме действия ферментов дает теория кислотно-основного катализа. Любая реакция, идущая с разрывом ковалентных связей, предполагает участие двух противоположных по характеру электронных компонентов. Электроны разрываемой связи должны оттягиваться к электро-фильному компоненту и уходить от нуклеофильного. Реагенты, которые могли бы обусловить такую электронную перестройку - это кислота и основание. Однако в одном и том же растворе создать одновременно высокие концентрации обоих компонентов невозможно, поскольку они нейтрализуют друг друга. В белковой молекуле фермента благодарязакреплению на каталитической площадке электрофильных и нуклеофильных групп не происходит прямой реакции нейтрализации. Это, собственно, и определяет акт катализа. Находясь на определенном расстоянии друг от друга, электрофильные и нуклеофильные группы каталитического участка фермента не только связываются с реагирующими группами субстрата, но и оказывают сильное поляризующее действие на группы субстрата. К этому следует добавить возможность флуктуации зарядов в комплексе ЕS, которая создает высокую степень эффективности данной поляризации. Это и является причиной снижения величины энергии активации при ферментативном катализе.
В соответствии с теорией ковалентного катализа некоторые ферменты взаимодействуют со своими субстратами, образуя нестабильные, ковалентно связанные фермент-субстратные комплексы. Из этих комплексов в ходе последующей реакции образуются продукты реакции, причем значительно быстрее, чем в случае некатализируемых реакций.
Таким образом, третья стадия, завершающаяся образованием продуктов реакции, обеспечивается процессами, протекающими на предыдущих стадиях.
Ферментативная кинетика изучает влияние различных факторов (концентрация S и E, рН, температура, давление, ингибиторы и активаторы) на скорость ферментативных реакций. Главной целью изучения кинетики ферментативных реакций является получение информации, позволяющей глубже понять механизм действия ферментов.
Кинетическая кривая позволяет определить начальную скорость реакции V 0 .
Кривая субстратного насыщения.
Зависимость скорости реакции от концентрации фермента.
Зависимость скорости реакции от температуры.
Зависимость скорости реакции от рН.
|
|
Оптимум рН действия большинства ферментов лежит в пределах физиологических значений 6,0-8,0. Пепсин активен при рН 1,5-2,0, что соответствует кислотности желудочного сока. Аргиназа, специфичный фермент печени, активен при 10,0. Влияние рН среды на скорость ферментативной реакции связывают с состоянием и степенью ионизации ионогенных групп в молекуле фермента и субстрата. Этот фактор определяет конформацию белка, состояние активного центра и субстрата, формирование фермент-субстратного комплекса, собственно процесс катализа. |

Математическое описание кривой субстратного насыщения, константа Михаэлиса .
|
|
Уравнение, описывающее кривую субстратного насыщения, было предложено Михаэлисом и Ментон и носит их имена (уравнение Михаэлиса-Ментен): V = (V MAX *[ S ])/(Km +[ S ]) , где Km – константа Михаэлиса. Легко рассчитать, что при V = V MAX /2 Km = [S], т.е. Km – это концентрация субстрата, при которой скорость реакции составляет ½ V MAX . С целью упрощения определения величины V MAX и Km уравнение Михаэлиса-Ментен можно пересчитать. 1/V = (Km+[S])/(V MAX *[S]), 1/V = Km/(V MAX *[S]) + 1/V MAX , |
|
|
1/ V = Km / V MAX *1/[ S ] + 1/ V MAX уравнение Лайнуивера-Берка. Уравнение, описывающее график Лайнуивера-Берка – это уравнение прямой линии (y = mx + c), где 1/V MAX – это отрезок, отсекаемый прямой на оси ординат; Km/V MAX - тангенс угла наклона прямой; пересечение прямой с осью абсцисс дает величину 1/Km. График Лайнуивера-Бэрка позволяет определить Km по относительно небольшому числу точек. Этот график также используют при оценке действия ингибиторов, о чем будет сказано ниже. Значение Km изменяются в широких пределах: от 10 -6 моль/л для очень активных ферментов, до 10 -2 – для малоактивных ферментов. |
Оценки Km имеют практическую ценность. При концентрациях субстрата в 100 раз превышающих Km, фермент будет работать практически с максимальной скоростью, поэтому максимальная скорость V MAX будет отражать количество присутствующего активного фермента. Это обстоятельство используют для оценки содержания фермента в препарате. Кроме того, Km является характеристикой фермента, что используется для диагностики энзимопатий.
Ингибирование активности ферментов.
Чрезвычайно характеристикой и важной особенностью ферментов является их инактивация под влиянием определенных ингибиторов.
Ингибиторы – это вещества, вызывающие частичное или полное торможение реакций, катализируемых ферментами.
Ингибирование ферментативной активности может быть необратимым или обратимым, конкурентным или неконкрентным.
Необратимое ингибирование – это стойкая инактивация фермента, возникающая в результате ковалентного связывания молекулы ингибитора в активном центре или в другом особом центре, изменяющим конформацию фермента. Диссоциация столь устойчивых комплексов с регенерацией свободного фермента практически исключена. Для преодоления последствий такого ингибирования организм должен синтезировать новые молекулы фермента.
Обратимое ингибирование – характеризуется равновесным комплексообразованием ингибитора с ферментом за счет нековалентных связей, вследствие чего такие комплексы способны к диссоциации с восстановлением активности фермента.
Классификация ингибиторов на конкурентные и неконкурентные основана на том, ослабляется (конкурентное ингибирование ) или не ослабляется (неконкурентное ингибирование ) их ингибирующие действие при повышении концентрации субстрата.
Конкурентные ингибиторы – это, как правило, соединения, структура которых сходна со структурой субстрата. Это позволяет им связываться в том же активном центре, что и субстраты, препятствуя взаимодействию фермента с субстратом уже на стадии связывания. После связывания ингибитор может быть превращен в некий продукт или остается в активном центре, пока не произойдет диссоциация.
Обратимое конкурентное ингибирование можно представить в виде схемы:
E↔ E-I → E + P 1
S (неакт)
Степень ингибирования фермента определяется соотношением концентраций субстрата и фермента.
Классическим примером подобного типа ингибирования является торможение активности сукцинатдегидрогеназы (СДГ) малатом, который вытесняет сукцинат из субстратного участка и препятствует его превращению в фумарат:

Ковалентное связывание ингибитора в активном центре приводит к инактивации фермента (необратимое ингибирование). Примером необратимого конкурентного ингибирования может служить инактивация триозофосфатизомеразы 3-хлорацетолфосфатом. Этот ингибитор является структурным аналогом субстрата – диоксиацетонфосфата и необратимо присоединяется к остатку глутаминовой кислоты в активном центре:
Некоторые ингибиторы действуют менее избирательно, взаимодействуя с определенной функциональной группой в составе активного центра разных ферментов. Так, связывание йодацетата или его амида с SH-группой аминокислоты цистеина, находящийся в активном центре фермента и принемающей участие в катализе, приводит к полной утрате активности фермента:
R-SH + JCH 2 COOH → HJ + R-S-CH 2 COOH
Поэтому эти ингибиторы инактивируют все ферменты, которые имеют SH-группы, участвующие в катализе.
Необратимое ингибирование гидролаз при действии нервно-паралитических газов (зарин, зоман) обусловлено их ковалентным связыванием с остатком серина в активном центре.
Метод конкурентного ингибирования нашел широкое применение в медицинской практике. Сульфаниламидные препараты – антагонисты п-аминобензойной кислоты, могут служить примером метаболизируемых конкурентных ингибиторов. Они связываются с дигидроптератсинтетазой – бактериальным ферментом, осуществляющим превращение п-аминобензоата в фолиевую кислоту, необходимую для роста бактерий. Бактерия погибает в результате того, что связавшийся сульфаниламид превращается в другое соединение и фолиевая кислота не образуется.
Неконкурентные ингибиторы обычно связываются с молекулой фермента в участке, отличном от места связывания субстрата, и субстрат непосредственно не конкурирует с ингибитором. Поскольку ингибитор и субстрат связываются с разными центрами возможно образование как комплекса E-I, так и комплекса S-E-I. Комплекс S-E-I тоже распадается с образованием продукта, однако с меньшей скоростью, чем E-S, поэтому реакция будет замедляться, но не остановится. Таким образом, могут протекать следующие параллельные реакции:
E↔ E-I ↔ S-E-I → E-I + P
Обратимое неконкурентное ингибирование встречается сравнительно редко.
Неконкурентные ингибиторы называют аллостерическими в отличие от конкурентных (изостерических ).
Обратимое ингибирование может быть количественно изучено на основе уравнения Михаэлиса-Ментен.
При конкурентном ингибировании V MAX остается постоянной, а Km возрастает.
|
|
|
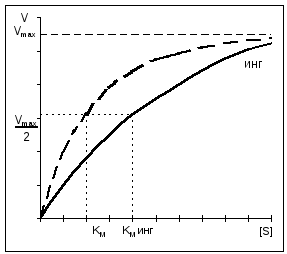
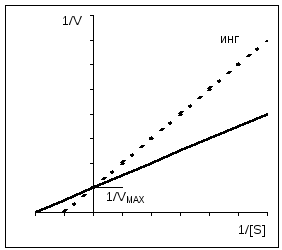
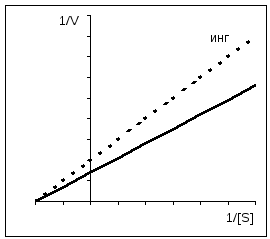
При неконкурентном ингибировании снижается V MAX при неизменном Km.
|
|
|

Если продукт реакции ингибирует фермент, катализирующий его образование, такой способ ингибирования называется ретроингибированием или ингибированием по принципу обратной связи . Например, глюкоза тормозит глюкозо-6-фосфатазу, которая катализирует гидролиз глюкозо-6-фосфата.
Биологическое значение такого ингибирования – регуляция определенных метаболических путей (см. следующее занятие).
ПРАКТИЧЕСКАЯ ЧАСТЬ
Задание студентам
1. Изучить денатурацию белков под действием растворов минеральных и органических кислот и при нагревании.
2. Обнаружить кофермент НАД в дрожжах.
3. Определить амилазную активность в моче (сыворотке крови).
9. ЭТАЛОНЫ ОТВЕТОВ НА ЗАДАЧИ , тестовые вопросы, используемые при контроле знаний на занятии (можно в виде приложения)
10. ХАРАКТЕР И ОБЪЕМ ВОЗМОЖНОЙ УЧЕБНО-ИССЛЕДОВАТЕЛЬСКОЙ РАБОТЫ ПО ТЕМЕ
(Указать конкретно характер и форму УИРС: подготовка реферативных выступлений, проведение самостоятельных исследований, имитационная игра, оформление истории болезни с использованием монографической литературы и др. формы)
Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
РЕФЕРАТ
Ферменты и ферментные реакции
Ферменты
Ферментативная активность
Реакционная и субстратная специфичность
Классы ферментов
Ферментативный катализ
Кинетика ферментативных реакций
Ингибиторы
Ферментативный анализ
свойство реакция катализ фермент
Ферменты
Ферменты являются биокатализаторами , т.е. веществами биологического происхождения, ускоряющими химические реакции. Организованная последовательность процессов обмена веществ возможна при условии, что каждая клетка обеспечена собственным генетически заданным набором ферментов. Только при этом условии осуществляется согласованная последовательность реакции. Ферменты принимают участие также в регуляции многих метаболических процессов, обеспечивая тем самым соответствие обмена веществ измененным условиям. Почти все ферменты являются белками . Известны также каталитически активные нуклеиновые кислоты -- «рибозимы».
Ферментативная активность
Каталитическое действие фермента, т. е. его активность , определяют в стандартных условиях по увеличению скорости (фиолетовый цвет на схеме) каталитической реакции (оранжевый цвет) по сравнению с некаталитической (желтый цвет). Обычно скорость реакции указывают как изменение концентрации субстрата или продукта за единицу времени (моль/(л·с)) . Так как каталитическая активность не зависит от объема раствора, в котором протекает реакция, активность фермента выражают в каталах ; 1 кат -- это количество фермента, которое превращает 1 моль субстрата за 1 с. Другой единицей активности является международная единица (E) -- количество фермента, превращающего 1 мкмоль субстрата в 1 мин (1 E = 16,7 нкат).
Реакционная и субстратная специфичность
Действие большинства ферментов высоко специфично . Понятие специфичности относится не только к типам каталитических реакций (реакционная специфичность ), но и к природе соединений - субстратов (субстратная специфичность ). В качестве примера на схеме приведены ферменты, расщепляющие химическую связь. Высокоспецифичные ферменты (тип А -- верхняя строка таблицы) катализируют расщепление только одного типа связи в субстратах определенной структуры. Ферменты типа Б (средняя строка) обладают ограниченной реакционной специфичностью, но широкой субстратной специфичностью. Ферменты типа В (с низкой реакционной и низкой субстратной специфичностями; нижняя строка) встречаются редко.
Классы ферментов
На сегодняшний день известно примерно 2000 различных ферментов. Разработанная система классификации учитывает реакционную и субстратную специфичности ферментов. Все ферменты включены в «Каталог ферментов» под своим классификационным номером (КФ), состоящим из четырех цифр. Первая цифра указывает на принадлежность к одному из шести главных классов . Следующие две определяют подкласс и подподкласс, а последняя цифра -- номер фермента в данном под подклассе. Например, лактатдегидрогеназа имеет номер КФ 1.1.1.27 (класс 1, оксидоредуктазы; подкласс 1.1, донор электрона -- СН-ОН; подподкласс 1.1.1, акцептор -- НАДФ + .)
В каждом из шести главных классов объединены ферменты, обладающие одинаковой реакционной специфичностью. О ксидоредуктаз ы (класс 1) катализируют окислительно-восстановительные реакции. Трансферазы (класс 2) переносят ту или иную функциональную группу от одного субстрата на другой. Для оксидоредуктаз и трансфераз необходим общий кофермент. Гидролазы (класс 3) также участвуют в переносе групп, однако акцептором здесь всегда является молекула воды. Лиазы (класс 4, называемые иногда «синтезами») катализируют расщепление или образование химических соединений, при этом образуются или исчезают двойные связи.
Изомеразы (класс 5) перемещают группы в пределах молекулы безизменение общей формулы субстрата. Лигазы («синтезы», класс 6) катализируют энергозависимые реакции присоединения и поэтому их действие Сопряжено с гидролизом нуклеозидтрифосфата (чаще всего АТФ).
Ферментативный катализ
Ферменты -- высокоэффективные катализаторы . Они повышают скорость катализируемой реакции в 10 12 раз и более. Для понимания механизма ферментативного катализа полезно прежде всего рассмотреть протекание некаталитической реакции.
Некаталитическая реакция (в отсутствие фермента)
В качестве примера рассмотрим реакцию типа А + В > С + D. Вещества A и Вв растворе окружены оболочкой из молекул воды (гидратной оболочкой) и под действием теплового движения перемещаются случайным образом. Они могут вступать в реакцию друг с другом только в том случае, когда сталкиваются в благоприятной ориентации, что маловероятно и происходит редко.
Для образования продуктов C + D комплекс A--В, возникший в результате соударения молекул, должен образовать переходное состояние , для чего требуется, как правило, значительная энергия активации E a . Поскольку получить эту энергию может только небольшая часть комплексов A--В, достижение переходного состояния -- еще более редкий случай, чем возникновение комплекса. В растворе большая часть анергии активации расходуется на преодоление гидратных оболочек между A и В, сближение реагентов и другие химические процессы , в которых эти реагенты участвуют. В результате в отсутствие катализатора образование продуктов происходит крайне редко и скорость реакции v незначительна, даже когда реакция термодинамически допустима, т. е. ДG < 0
Ферментативная реакция
Ферменты специфически связывают реагенты (свои субстраты) в активном центре . При этом субстраты ориентируются таким образом, что приобретают оптимальное положение для образованияпереходного состояния (1-3). Сближение и необходимая ориентации реагентов значительно повышают вероятность образования продуктивного комплекса A--B. Кроме того, связывание субстрата в активном центре приводит к удалению гидратной оболочки субстрата.В результате удаления молекул воды в активном центре фермента во время катализа создаются совершенно другие условия, чем в растворе (3-5). Еще одним важным фактором является вследствие взаимодействия между аминокислотными остатками белка и субстратом (4). Таким образом, переходное состояние в случае ферментативной реакции требует меньшей энергии активации. Кроме того, многие ферменты во время катализа переносят специфические группировки с субстрата или на субстрат. Особенно часто осуществляется перенос протонов. Этот ферментативный кислотно-основной катализ значительно более эффективен, чем обмен протонов с кислотами и основаниями в растворе. Часто химические группировки ковалентно присоединяются к остаткам фермента. Это явление называют ковалентным катализом .. Основы ферментативного катализа
Несмотря на то, что сегодня трудно количественно оценить вклад отдельных каталитических эффектов, решающим фактором считается стабилизация переходного состояния в активном центре фермента. При этом наиболее существенным моментом является прочное связывание не столько субстрата, сколько его переходного состояния. Данное положение подтверждается крайне высоким сродством многих ферментов по отношению к аналогам переходного состояния, что можно пояснить простой механической аналогией (на схеме справа): если хотят перекатить металлические шарики (реагенты) с места EA (состояние субстрата) в энергетически более высокое переходное состояние, а затем в EP (состояние продукта), нужно расположить магнит (катализатор) таким образом, чтобы сила притяжения действовала не на EA (а), а на переходное состояние (б).
Кинетика ферментативных реакций
Кинетика ферментативной реакции (т. е. зависимость скорости реакции от ее условий) определяется в первую очередь свойствами катализатора , вследствие чего она значительно сложнее, чем кинетика некаталитических реакций
Модель Михаэлиса-Ментен
Полный математический анализ ферментативной реакции приводит к сложным уравнениям, не пригодным для практического применения. Наиболее удобной оказалась простая модель, разработанная в 1913 г. Она объясняет характерную гиперболическую зависимость активности фермента от концентрации субстрата (1) и позволяет получать константы, которые количественно характеризуют эффективность фермента.
Модель Михаэлиса-Ментен исходит из того, что вначале субстрат А образует с ферментом E (З) комплекс, который превращается в продукт В намного быстрее, чем в отсутствие фермента. Константа скорости k кат (2) намного выше, чем константа некаталитической реакции k. Константу k кат называют еще «числом оборотов » поскольку она соответствует числу молекул субстрата, превращаемых в продукт одной молекулой фермента за 1 с. Согласно этой модели, активность фермента определяется долей комплекса EA от общей концентрации фермента [E] t , т. е., отношением / [E] t (З). С целью упрощения модель предполагает, что E, А и ЕА находятся в химическом равновесии согласно закону действующих масс, что дает в итоге для диссоциации комплекса EA уравнение:
[E][A]/ = K m Поскольку [E] t = [E] + ,
= [E] t [А]/(К m + [А])
Из v = k кат (2) и предыдущего выражения получают уравнение Михаэлиса-Ментен (4).
Уравнение содержит две величины (два параметра ), которые не зависят от концентрации субстрата [A], но характеризуют свойства фермента: это произведениеk кат [E] t , соответствующее максимальной скорости реакции V при высокий концентрации субстрата, и константа МихаэлисаК m , характеризующая сродство фермента к субстрату. Константа Михаэлиса численно равна той концентрации субстрата [A].при которой н достигает половины максимальной величины V (если v = V/2, то [A] / (К m + [A]) = 1/2, т. е. K m = [А]). Высокое сродство фермента к субстрату характеризуется низкой величиной К m и наоборот,
Модель Михаэлиса-Ментен основывается на нескольких не совсем реальных допущениях, таких, как необратимое превращение EA в E + В, достижение равновесия между E, A и EA, отсутствие в растворе других форм фермента, кроме E и EA. Только при соблюдении этих гипотетических условий K m соответствует константе диссоциации комплекса, а k кат -- константе скорости peакции EA > E + В.
Определение V и К m
В принципе V и К m можно определить по графику зависимости v от [A] (рис. слева). Так как v асимптотически достигает V с возрастанием концентрации субстрата [A], то затруднительно получить надежную величину V и К m (рис. слева) путем экстраполяции.
Для удобства расчетов уравнение Михаэлиса-Ментен можно преобразовать так, чтобы экспериментальные точки лежали на прямой. При одном из таких графических преобразований в так называемом графике Иди-Хофсти (pиc. справа) строят график зависимости v от v/[A]. В этом случае точка пересечения прямой, полученной путем наилучшей линейной аппроксимации экспериментальных точек, с осью ординат соответствует V, а тангенс угла наклона равен -K m . Такой графический подход дня определения V и К m также не оптимален. В настоящее время данные ферментативной кинетики обрабатывают быстрее и более объективно с помощью вычислительной техники.
Ингибиторы
Многие соединения могут влиять на обмен веществ, модулируя активность соответствующих ферментов. Особенно важные функции при этом выполняют ингибиторы ферментов . Ингибиторами ферментов являются многие лекарственные вещества природного или синтетического происхождения. Метаболиты также могут быть ингибиторами ферментов в процессах регуляции.
Типы ингибирования
Большинство ингибиторов ферментов действуют обратимо , т. е. не вносят в молекулу фермента каких-либо изменений после своей диссоциации. Однако существуют также необратимые ингибиторы ферментов, которые необратимо модифицируют целевой фермент. Принцип действия ингибитора, тип его ингибирования определяют путем сравнения кинетики реакции в присутствии ингибиторам без него (см. схему Б). Различают конкурентное (А, слева) и неконкурентное (А, справа) ингибирование . В регуляции обмена веществ важную роль играет аллостерическое ингибирование (А, 6).
Так называемые аналоги субстрата (2) имеют свойства, подобные свойствам субстрата целевого фермента. Они обратимо блокируют часть молекул имеющегося в наличии фермента, но не могут далее превращаться в продукт. Поэтому для достижения половины максимальной скорости реакции необходимы более высокие концентрации субстрата: в присутствии такого ингибитора константа МихаэлисаK m растет (Б). Субстрат в высоких концентрациях вытесняет ингибитор с фермента. Поэтому максимальная скорость V при этом типе торможения не претерпевает изменений. Так как субстрат и ингибитор конкурируют за место связывания на ферменте, данный тип торможения называют конкурентным. Аналоги переходного состояния (3) также действуют как конкурентные ингибиторы.
Если ингибитор реагирует с функционально важной группой фермента, не препятствуя связыванию субстрата, такое ингибирование называется неконкурентным (на схеме справа). В этом случае K m остается неизменной, напротив уменьшается концентрация функционально активного фермента [Е] t и, следовательно, максимальная скорость реакции V. Неконкурентные ингибиторы действуют как правило необратимо , поскольку они модифицируют функциональные группы целевого фермента (4).
В случае так называемых "суицидныхсубстратов " (5) речь идет о субстратных аналогах, содержащих дополнительно реакционную группу. Вначале они связываются обратимо, а затем образуют ковалентное соединение с активным центром фермента. Поэтому ингибирование такими соединениями проявляется как неконкурентное. Известным примером такого ингибитора является антибиотик пенициллин .
Аллостерические ингибиторы связываются с отдельными участками фермента вне активного центра (6). Такое связывание влечет за собой конформационные изменения в молекуле фермента, которые приводят к уменьшению его активности. Аллостерические эффекты встречаются практически только в случае олигомерных ферментов. Кинетику таких систем нельзя описать с помощью простой модели Михаэлиса-Ментен.
Кинетика ингибирования
Конкурентное ингибирование легко можно отличить от неконкурентного при использовании графика Иди-Хофсти . Как уже упоминалось, конкурентные ингибиторы влияют только на K m , но не на V. Полученные в отсутствие и в присутствии ингибитора прямые на графике пересекаются на оси ординат. Прямые для неконкурентного ингибирования имеют одинаковый наклон (К m не изменяется), однако по мере увеличения концентрации ингибитора отрезки, отсекаемые этими прямыми на оси ординат, становятся все короче. Для аллостерических ферментов нельзя применять график Иди-Хофсти, имеющий в этом случае нелинейный характер (здесь не приведен).
Ферментативный анализ
Ферменты играют важную роль в биохимическом анализе . В биологических материалах, например в жидкостях организма, с помощью определения каталитической активности можно обнаружить ферменты в ничтожно малых концентрациях. Ферменты можно использовать как реагенты для определения концентраций метаболитов, например уровня глюкозы в крови (схема В). В большинстве ферментативных анализов применяется фотометрия.
Основы спектрофотометрии
Многие молекулы поглощают свет в видимой или ультрафиолетовой области спектра. Это свойство можно использовать для определения концентраций. Величина поглощения зависит от типа и концентрации вещества, а также от длины волны используемого света. Поэтому применяют монохроматический свет , т. е. свет определенной длины волны, который можно выделить из белого света с помощью монохроматора . Монохроматический свет интенсивности I 0 проходит через прямоугольную ячейку из стекла или кварца (кювету), в которой находится раствор поглощающего вещества. Интенсивность I выходящего света, ослабленного поглощением, измеряется с помощью детектора. Поглощение света (А) раствора (оптическая плотность ) определяется как отрицательный логарифм отношения I / I 0 . Закон Ламберта-Бера гласит, что А пропорциональна концентрации (с) вещества и толщине (d) слоя раствора. Коэффициент экстинкции е зависит, как было указано выше, от типа вещества и длины волны.
Определение активности лактатдегидрогеназы
Определение активности лактатдегидрогеназы [ЛДГ (LDH)] основано на том факте, что восстановленный кофермент НАДН + H + поглощает свет при 340 нм, в то время как у НАД + (NAD +) при этой длине волны поглощение отсутствует. Спектры поглощения (т. е. графики зависимости А от длины волны) субстрата и кофермента в ЛДГ-реакции показаны на рис. Б1.
Различия в поглощении НАД + и НАДН между 300 и 400 нм обусловлены изменениями никотинамидного кольца при окислении или восстановлении.
Для определения активности в кювету помещают, прежде всего растворы лактата и НАД + и регистрируют поглощение при постоянной длине волны 340 нм. Некаталитическая реакция протекает с очень низкой скоростью. Поэтому измеряемые количества НАДН образуются только после добавления ЛДГ. Так как скорость увеличения поглощения ДA/Дt по закону Ламберта-Бера пропорциональна скорости реакции Дc/Дt , активность ЛДГ можно рассчитать с помощью коэффициента экстинкции е при 340 нм или путем сравнения со стандартным раствором.
Ферментативное определение глюкозы
Большинство биомолекул не поглощают свет в видимой или ультрафиолетовой областях спектра. Кроме того, они обычно присутствуют в смеси с другими соединениями, которые также дают аналогичные химические реакции.
Обе трудности можно преодолеть с помощью подходящего фермента для избирательного превращения определяемого метаболита в окрашенное вещество, которое далее определяют по интенсивности поглощения света.
Обычный метод определения глюкозы в крови основан на двух последовательных реакциях:
1) образование глюконолактона и пероксида водорода H 2 O 2 под действием фермента глюкозооксидазы ;
2) окисление бесцветного вещества пероксидом водорода в окрашенное зеленое соединение в реакции, катализируемой пероксидазой.
Когда вся имеющаяся в пробе глюкоза израсходована, количество образованного окрашенного вещества можно определить по светопоглощению, которое прямо пропорционально первоначальному содержанию глюкозы.
Размещено на Allbest.ru
Подобные документы
Ускорение химических реакций с помощью катализаторов. Особенности ферментов (энзимов) как высокоспецифичных белков, выполняющих функции биологических катализаторов. Строение ферментов, их специфичность и классификация. Этапы ферментативного катализа.
презентация , добавлен 20.11.2014
Классификация ферментов, их функции. Соглашения о наименовании ферментов, структура и механизм их действия. Описание кинетики односубстратных ферментативных реакций. Модели "ключ-замок", индуцированного соответствия. Модификации, кофакторы ферментов.
презентация , добавлен 17.10.2012
Характеристика ферментов, органических катализаторов белковой природы, которые ускоряют реакции, необходимые для функционирования живых организмов. Условия действия, получение и применение ферментов. Болезни, связанные с нарушением выработки ферментов.
презентация , добавлен 19.10.2013
Общая характеристика и основные типы ферментов. Химические свойства ферментов и катализируемых ими реакций. Селективность и эффективность ферментов. Зависимость от температуры и от среды раствора. Активный центр фермента. Скорость ферментативных реакций.
презентация , добавлен 06.10.2014
Определение ферментов как специфических белков, присутствующих во всех живых клетках биологических катализаторов. Пространственность структурной молекулы ферментов, процесс биосинтеза оксидоредуктазы, трансферазы, гидролазы, лиазы, изомеразы и лигазы.
контрольная работа , добавлен 27.01.2011
Кинетические исследования ферментативных реакций для определения ферментов и сравнения их скоростей. Образование из фермента и субстрата фермент-субстратного комплекса за счет сил физической природы. Факультативные организмы, автотрофы и гетеротрофы.
контрольная работа , добавлен 26.07.2009
Ферменты: биохимическое строение и физиологическая роль. Анализ методики определения активности ферментов и ферментативного спектра в жидкостях организма. Основные ферменты в моче в норме и при патологии. Ферментный спектр мочи при заболеваниях почек.
доклад , добавлен 10.03.2015
Изучение ферментов, их свойств и механизма биологического действия. Проведение исследования современных представлений о механизме ферментативного трансаминирования. Разработка общей теории пиридоксалевого катализа. Строение фермент-субстратного комплекса.
реферат , добавлен 14.03.2015
Специфические белки, катализирующие химические реакции в живых системах. Характеристика и классификация ферментов, их размеры и строение. Влияние условий среды на активность ферментов: факторы и кофакторы; заболевания, связанные с нарушением их выработки.
презентация , добавлен 07.05.2015
Изучение назначения ферментов или энзимов - белковых молекул или молекул РНК (рибозимов) или их комплексов, ускоряющих (катализирующих) химические реакции в живых системах. Локализация ферментов в клетке. Наследственные и приобретенные ферментопатии.
Похожие статьи
-
Этногенез и этническая история русских
Русский этнос - крупнейший по численности народ в Российской Федерации. Русские живут также в ближнем зарубежье, США, Канаде, Австралии и ряде европейских стран. Относятся к большой европейской расе. Современная территория расселения...
-
Людмила Петрушевская - Странствия по поводу смерти (сборник)
В этой книге собраны истории, так или иначе связанные с нарушениями закона: иногда человек может просто ошибиться, а иногда – посчитать закон несправедливым. Заглавная повесть сборника «Странствия по поводу смерти» – детектив с элементами...
-
Пирожные Milky Way Ингредиенты для десерта
Милки Вэй – очень вкусный и нежный батончик с нугой, карамелью и шоколадом. Название конфеты весьма оригинальное, в переводе означает «Млечный путь». Попробовав его однажды, навсегда влюбляешься в воздушный батончик, который принес...
-
Как оплатить коммунальные услуги через интернет без комиссии
Оплатить услуги жилищно-коммунального хозяйства без комиссий удастся несколькими способами. Дорогие читатели! Статья рассказывает о типовых способах решения юридических вопросов, но каждый случай индивидуален. Если вы хотите узнать, как...
-
Когда я на почте служил ямщиком Когда я на почте служил ямщиком
Когда я на почте служил ямщиком, Был молод, имел я силенку, И крепко же, братцы, в селенье одном Любил я в ту пору девчонку. Сначала не чуял я в девке беду, Потом задурил не на шутку: Куда ни поеду, куда ни пойду, Все к милой сверну на...
-
Скатов А. Кольцов. «Лес. VIVOS VOCO: Н.Н. Скатов, "Драма одного издания" Начало всех начал
Некрасов. Скатов Н.Н. М.: Молодая гвардия , 1994. - 412 с. (Серия "Жизнь замечательных людей") Николай Алексеевич Некрасов 10.12.1821 - 08.01.1878 Книга известного литературоведа Николая Скатова посвящена биографии Н.А.Некрасова,...